Поиск по сходству. Нуклеотидный Blast
Задание 1. Таксономия и функция последовательности из практикума 6.
В практикуме 6 мы учились с помощью прямой и обратной хроматограммы анализировать секвенированные
последовательности. С помощью команды consambig пакета EMBOSS
была получена
консенсусная последовательность. Организм не был известен.
Для того, чтобы узнать, кому принадлежит данная последовательность, был использован алгоритм BLASTN
со стандартными параметрами. (Поиск в банке - Nucleotide collection.)
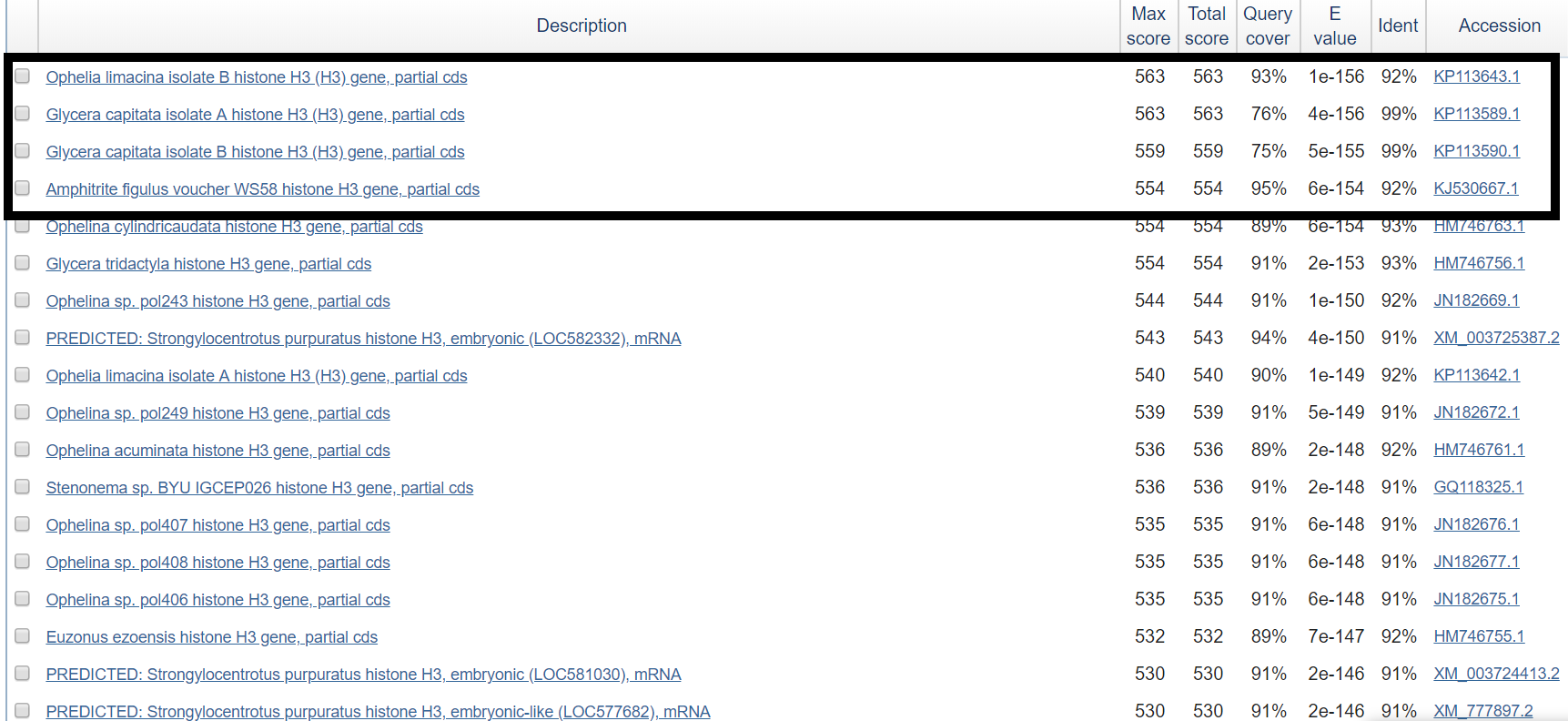
Как видно из поиска, полученная последовательность кодирует части гистона H3. Последовательности, обведенные рамочкой, были получены
в МГУ им. М.В.Ломоносова и принадлежат кольчатым червям.
Общее систематическое положение для первых четырех находок
Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Protostomia; Lophotrochozoa; Annelida; Polychaeta;
Очень трудно сделать однозначный вывод об исследуемой последовательности. Лучший E-value у первой находки. При этом у нее
не лучший процент совпадений (поле Indent). У второй последовательности в поиске такой же Score, как у первой, лучше процент
сопадений, и нет отличия в порядке E-value, хотя и страдает процент покрытия (поле Query cover)
Построим множественное выравнивание первых четырех последовательностей и исследуемой последовательности.

Последовательности на рисунке выше расположены в том порядке, как их предлагает поиск в blastn. Первая последовательность
- полученная с помощью секвенатора.
Видно,что лучше всего выравниваются последовательности, принадлежащие виду Glycera capitata (последовательности
2 и 3, не считая исследуемой). Там почти нет гэпов, нет подвижек и замен. Последовательность 1 (вид Ophelia limacina)
выравнивается будучи комплементарной и обратной, соответственно. Но все равно видно, что присутствуют замены С на Т (позиции 201, 240),
и ряд других. Последовательность 4 (вид Amphitrite figulus) тоже присутствуют замены, хоть и в меньшем количестве.
Так же стоит заметить, что четверая в выдаче BLAST последовательность принадлежит организму Amphitrite figulus. В записи
этой последовательности можно увидеть в поле TITLE - Genetic revision of the White Sea Kandalaksha Bay invertebrate fauna, и в поле
Journal - указания на факультет Биоинженерии и Биоинформатики. Это дает основания полагать, что исследуемая последовательность
принадлежит Amphitrite figulus.
Многие несовпадения с консенсусной последовательностью обусловлены
не очень четкими данными, полученными в процессе секвенирования, и точный вывод сделать нельзя. Основываясь на вышесказанном,
я остановлюсь на cубклассе (subclass).
Систематическое положение объекта, которому принадлежит исследуемая последовательность:
Eukaryota; Metazoa; Lophotrochozoa; Annelida; Polychaeta;
Scolecida
Задание 2. Сравнение списков находок тремя разными видами BLAST
Таблица с выдачей Blast для последовательности из задания 1.
Для удобства сравнения количество записей в выдаче - 50. Исключение таксона было сделано для того, что посмотреть,
насколько похожи гистоны у выше стоящих представителей, и, конечно, в попытке ограничить выдачу.
Таблица сравнения для консенсусной последовательности из задания 1.
| Название алгоритма |
Параметры алгоритма |
Число находок |
| BlastN default |
Ограничение по таксону - исключен Scolecida (taxid:105387), остальные параметры по умолчанию. |
50 |
| BlastN sensitive |
Ограничение по таксону - исключен Scolecida (taxid:105387), все по умолчанию, кроме порога на E-value - 1e-140,
выбрана минимальная длина слова - 7. |
19 |
| MegaBlast |
Ограничение по таксону - исключен Scolecida (taxid:105387), все по умолчанию, кроме порога на E-value - 1e-130,
выбрана минимальная длина слова - 16. |
22 |
То, что было найдено BLAST default, включает в себя 2-ой и 3-ий поиски
целиком. Первым 11-ти находкам все три алгоритма дали одинаковый вес относительно друг друга - порядок
в котором их выдает поиск. Дальше начинаются разночтения.
Очень интересно, что меняется порядок, то есть вес алгоритмы считают по разному. Например, запись с ID - KX589037.1,
находится в первом поиске на
15 позиций ниже, чем в третьем поиске.
При этом вес в битах у них близок и процент схожести одинаковый. Но BlastN с параметрами по умолчанию находит большее количество более
похожих последовательностей.
Наибольший показатель E-value в третьем поиске, с помощью чувствительного MegaBlast - 4.25e-160, там же наивысший вес в битах
- 575. Максимальная длина выравнивания у всех трех запросов одинаковая - 395.
Можно сделать вывод, что похожие последовательности хорошо ищет MegaBlast, особенно когда он настроен более чувствительно. Хотя
и чувствительный BlastN нашел все то же самое, и даже меньше, немного иначе оценив по весу несколько записей. Это происходит
за счет того, что минимальная длина слова у BlastN - 7, а у MegaBlast - 16.
Так как в запросах не изменялись штрафы за гэпы и за совпадения\несовпадения, про них ничего нельзя сказать.
Таблица в Excel для всех трех поисков.
Для следующей части был взят ген rrn18 (18S ribosomal RNA) из митохондриального генома
Вельвичии Удивительной. В столбце Число находок указано количество найденных последовательностей,
в некоторых из которых существует несколько участков выравнивания.
Таблица сравнения для некодирующей РНК из предыдущего практикума.
| Название алгоритма |
Параметры алгоритма |
Число находок |
| BlastN default |
Ограничение по таксону: поиск в Acrogymnospermae (taxid:1437180), исключен Welwitschia (taxid:3376)
остальные параметры по умолчанию. |
50 записей, 260 выравниваний |
| BlastN sensitive |
Ограничение по таксону: поиск в Acrogymnospermae (taxid:1437180), исключен Welwitschia (taxid:3376),
все остальное по умолчанию, кроме порога на E-value - 1e-120, минимальная длина слова - 7. |
37 записей, 42 выравнивания |
| MegaBlast |
Ограничение по таксону: поиск в Acrogymnospermae (taxid:1437180), исключен Welwitschia (taxid:3376),
- длина слова - 16, порог на E-value - 1e-120 |
27 записей, 32 выраванивания |
 |
 |
 |
| BlastN default |
BlastN sensitive |
MegaBlast |
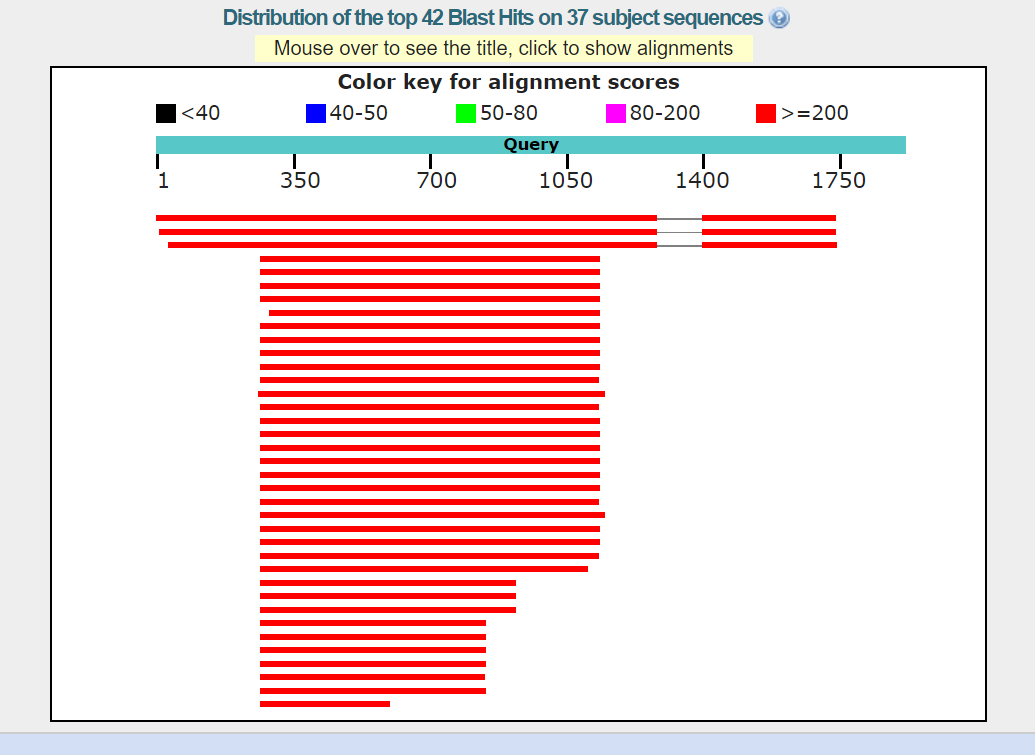
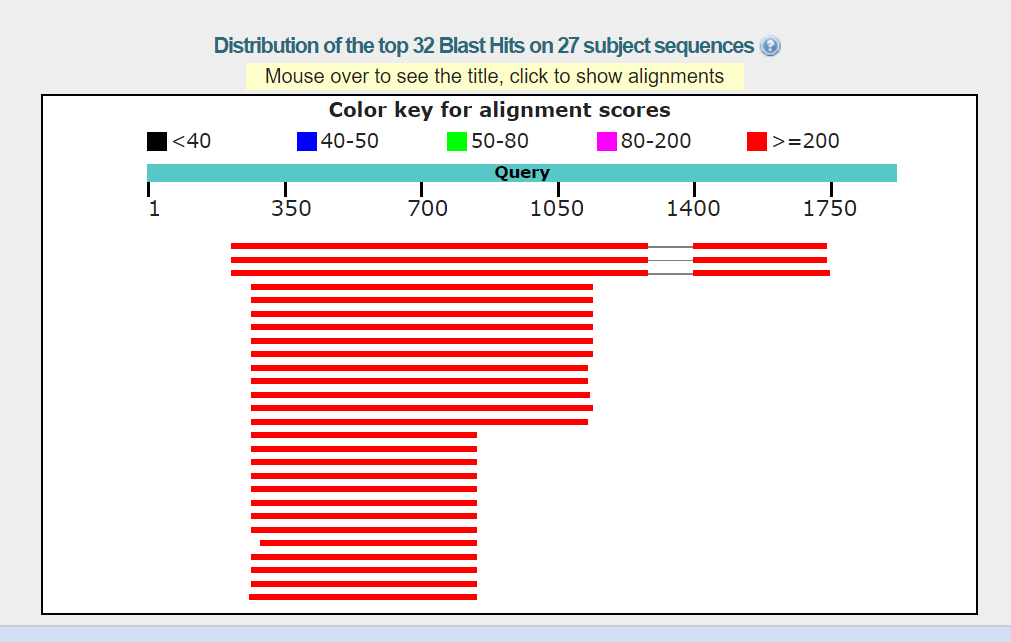
BlastN с параметрами по умолчанию включает в себя все находки из других поисков.
Совпадают первые три записи у всех трех поисков. Далее у BlastN обоих видов порядок находок
совпадает, а у MegaBlast меняется. Разные алгоритмы имеют по умолчанию разные
штрафы за гэпы.
Так же интересно заметить на картинках выше, BlastN со стандартными
параметрами ищет любые совпадения в последовательностях, поэтому выравнивание происходит "кусочками".
Таблица в Excel для всех трех поисков.
Задание 3. Поиск гомологов белков в неаннотированном геноме оргнизма Amoeboaphelidium protococcarum.
Использовался алгоритм tblastn. Был произведен поиск гомологов для белков TERT_SCHPO - теломераза,восстанавливающая
длину хромосом, EIF3G_SCHPO - один из факторов инициации трансляции, TBB_NEUCR - тубулин, участвующий в обраовании микротрубочек.
Для теломеразы получен следющий результат:
Length=988
Score E
Sequences producing significant alignments: (Bits) Value
scaffold-17 108 1e-23
unplaced-307 102 7e-22
scaffold-105 33.9 0.51
unplaced-647 28.1 4.9
Так как вес выравнивания довольно мал для такой длины выравнивания, можно сказать, что
у Amoeboaphelidium protococcarum отсутствуют гомологи данного белка.
Для фактора трансляции получен результат:
Length=282
Score E
Sequences producing significant alignments: (Bits) Value
scaffold-20 95.5 2e-21
scaffold-444 92.0 2e-20
scaffold-170 38.9 0.002
scaffold-17 37.4 0.008
scaffold-199 37.0 0.010
scaffold-212 35.8 0.023
scaffold-105 34.7 0.052
scaffold-96 33.5 0.12
scaffold-423 33.1 0.17
scaffold-287 32.3 0.30
scaffold-22 31.2 0.68
scaffold-57 29.6 2.1
Можно сказать, что есть некое частичное сходство белка и участка генома. Нельзя говорить о полной
гомологии. Ниже приведено выравнивание для первого в списке предпологаемого участка гомологии.
> scaffold-20
Length=45210
Score = 95.5 bits (236), Expect = 2e-21, Method: Compositional matrix adjust.
Identities = 109/287 (38%), Positives = 157/287 (55%), Gaps = 32/287 (11%)
Frame = -1
Query 20 PSIQTFDNPDGTKTMIEFRIDDNGkkvkvtrvirktvitERVQHAVaerkkwkkfgkeag 79
P I+ + DG K ++E+ +D G+K+K+TR ++ V + HAV ER+ WKKFG G
Sbjct 8619 PEIKVTEQ-DGLKVIVEYSTNDKGEKIKITRKVKTVVRK*KANHAVIERR*WKKFG*ATG 8443
Query 80 kNSGVDARTTSVGENVQLRLQLGWTTTkeeeqdeaalaaakvkakGSSVVRCRACKGNHF 139
+ D T++GE V L+L G E E VK +S + CR CKG HF
Sbjct 8442 HGT-FDEGITTLGEEVTLKL--GVRALNEVE*KPKLTVDE*VKKLTTSAITCRVCKGEHF 8272
Query 140 TAQCPYKSIIGPVDEPPLD--ASPVS------SRASGALGEK-----GRYIAPHLRA--- 183
TA+CPYK + P+ E LD ASP + S A GA G K G+YI P RA
Sbjct 8271 TAKCPYKDTLTPLSET-LDKLASPAAFSSDSVSGADGAAGAKPTLGGGKYIPPSQRAKMG 8095
Query 184 ------GSGRESG--DSMFKRERDDSA---TLRVTNlsddtreeelrdlfrrFGGIQRVY 232
+G SG S ++ D TLRV+NLS+ ++ +L LF +FGGI RV
Sbjct 8094 IPPSASAAGSTSGRPSSAYQPPGGDGGELTTLRVSNLSEYAQD*DLYSLFSKFGGI*RVK 7915
Query 233 LAKDKETGRAKGFAFVSYYDRDCAIKARDRLDGYGWNNLILRCEFSK 279
+A+D ET +G+AFV++ R A A +++DGY ++NLIL+ E+++
Sbjct 7914 VAQDYETRMCRGYAFVTFNIRRSAELAMEKMDGYPFDNLILKVEWAQ 7774
Вполне вероятно, что совпадение случайно, потому как длинных консервативных участков
выделить нельзя. Но вес довольно неплохой для длины белка, а так же есть совпадения в такиех аминокислотах,
как триптофан. Это позволяет сказать, что возможно это давно разошедшиеся частично гомологичные белки. О сохранении функций
говорить сложно.
Для одного из доменов тубулина была получен следующий результат.
Length=447
Score E
Sequences producing significant alignments: (Bits) Value
unplaced-665 742 0.0
scaffold-26 693 0.0
unplaced-5 348 1e-105
scaffold-57 348 1e-105
scaffold-423 161 6e-49
Можно говорить о гомологии. Большой вес, маленькое значение E-value. Этот домен тубулина имеется в геноме у
Amoeboaphelidium protococcarum.
Задание 4. Поиск гена белка в одном из контигов организма Amoeboaphelidium protococcarum
Был выбран контиг
unplaced-10 37804
В нем с помощью алгоритма BlastX был произведен поиск генов. Примерно с одинковыми значениями
E-value и процентом схожести оказались два белка - ABC - трасмембранный транспортер, состоящий из
двух доменов.
(RIA80538.1)
Более правдоподобным считать именно его. Потому что он первый в поиске, у выраванивания
контига с последовательностью этого белка - лучший вес, а еще он встречается в первых номерах
выдачи с процентом схожести (70%) пять раз. Выраванивание содержит всего 3 гэпа, и E-value равно 0.0.
Ниже приведено выравнивание с первой находкой.
Score Expect Method Identities Positives Gaps Frame
873 bits(2255) 0.0 Compositional matrix adjust. 433/621(70%) 508/621(81%) 3/621(0%) +1
Query 3988 SKSSARVGVNARFFA*MNKLVPILVPSVFCREsallvslaavliaRTWLDIWFSSFNGVV 4167
++ R+GV+ARF A + KL+PI +P + +ESALLVSLA VLIARTWLDIWFS FNG V
Sbjct 63 NRKQRRIGVDARFLAQIKKLLPICIPGIASKESALLVSLAMVLIARTWLDIWFSGFNGHV 122
Query 4168 VKSIVSRDRRAFVARAGVEFSLMMWPLSIVNNSLKMLINALSICFRDRLTTYAHEAYLSD 4347
VK+IVSRDR+ F+ARA VEF MMWP+SIVNNSLK+ I+AL++ FR RLT YAHE YL+D
Sbjct 123 VKAIVSRDRKTFIARAIVEFGFMMWPMSIVNNSLKLTISALALAFRTRLTRYAHEQYLND 182
Query 4348 ITFYKVSNIDNRI*NADQLLTQDIDKFSENLAHLYSDISKPLVDIFLFAIKLGQAIGGES 4527
ITFYK+SNIDNRI NADQLLTQDIDKF+ENL+HLYSDI+KP+VD+ LFA KLG+AIG E+
Sbjct 183 ITFYKISNIDNRIQNADQLLTQDIDKFAENLSHLYSDIAKPVVDMALFAYKLGEAIGREA 242
Query 4528 PLIMVSYFLASGVFLRVISPPFGKYTAQEQKLEGDFRFTHSRIITHAEEIAFYRGSEREK 4707
P M++YF SG LR ISPPFGK+TA EQKLEGDFRFTHSRIITH+EEIAFY G ERE+
Sbjct 243 PFYMIAYFFVSGALLRAISPPFGKFTAMEQKLEGDFRFTHSRIITHSEEIAFYGGGERER 302
Query 4708 TFVN*SFRRILEHLYKVNVLRFANGILDSVAVKYCATILAYWLLSRPVFDPRYATEQMGS 4887
VN SF +I+ H+ K+ LRFANGI DSV VKYCAT+ AY+LL+RPVFDP+YATE MG
Sbjct 303 AVVNGSFDKIVRHVKKIYRLRFANGIFDSVLVKYCATMTAYYLLARPVFDPKYATEFMGK 362
Query 4888 AGDDPTKIMEDYSRNSSYLVNLS*AVGRVILAGRDLTRFAGYTSRVAELFEVLEDVKRGR 5067
DPTKIMEDYSRNS YLVNLS AVGR+ILAGRDLTRFAGYTSRVAELF+VLEDV +GR
Sbjct 363 LDSDPTKIMEDYSRNSGYLVNLSQAVGRLILAGRDLTRFAGYTSRVAELFDVLEDVNKGR 422
Query 5068 YERSMV-NDTSGAQLTR-VE*SDLKGVIKKSDNADIDFDKVPIITPNGDVLIESLSINCK 5241
YER+MV ND++ A ++R V +DLKG + D I FDKVPIITPN D+L++ LS +
Sbjct 423 YERTMVNNDSNEANISRAVTPNDLKGKVITKDGV-IIFDKVPIITPNNDLLVKELSFKVE 481
Query 5242 HGTSVLVTGPNGSGKSSLFRILGELWPLFGGTLTKPEAS*FFYVPQKPYLCLGSLRDQLL 5421
G + L++GPNG GKSSLFRILG+LWPLF G +TKP AS FYVPQKPYL LG+LRDQ++
Sbjct 482 TGMNCLISGPNGCGKSSLFRILGDLWPLFDGVVTKPSASKLFYVPQKPYLALGTLRDQVI 541
Query 5422 YPHDKETALKNGATDDKLMELLKMVHLEYLVDREGGWDAVQDWADVLSGGEK*RMAMGRL 5601
YP K A DD+LMELL +VHL YL REGGWDAVQDWADVLSGGEK R+AM RL
Sbjct 542 YPDTKAQARSKDFNDDELMELLNVVHLGYLAKREGGWDAVQDWADVLSGGEKQRIAMARL 601
Query 5602 FYHKPLWAVLDECTSAVSIDVEGLMYTKAKELGITLFTVSHRTSLFKYHEYLLKFDGNGQ 5781
FYH+P +A+LDECTSAVS+DVEG+MYT A+ LGITLFTVSHR SL KYHE+LL+FDG G
Sbjct 602 FYHRPQFAILDECTSAVSVDVEGIMYTHARTLGITLFTVSHRHSLVKYHEWLLRFDGEGG 661
Query 5782 YEFSKLE*GDTSSPFAFTKLK 5844
YEF KLE GD ++PF F+ K
Sbjct 662 YEFRKLEEGDLTTPFTFSHGK 682
ABC (ATP-binding cassette) транспортер состоит
из 2 доменов и является трансмембранным интегральным белком, который активно
транспортирует лиганды через биологические мембраны с помощью энегрии АТФ. Это
довольно хорошо изученный белок. Некоторые его гомологи несут в себе и иные функции,
как, например, репарация ДНК.
Задание 5.Карта локального сходства геномов двух бактерий
Были выбраны две бактерии:
| Rickettsia prowazekii str. Madrid E (a-proteobacteria) |
NC_000963.1 |
| Rickettsia canadensis str. McKiel (a-proteobacteria) |
NC_009879.1 |
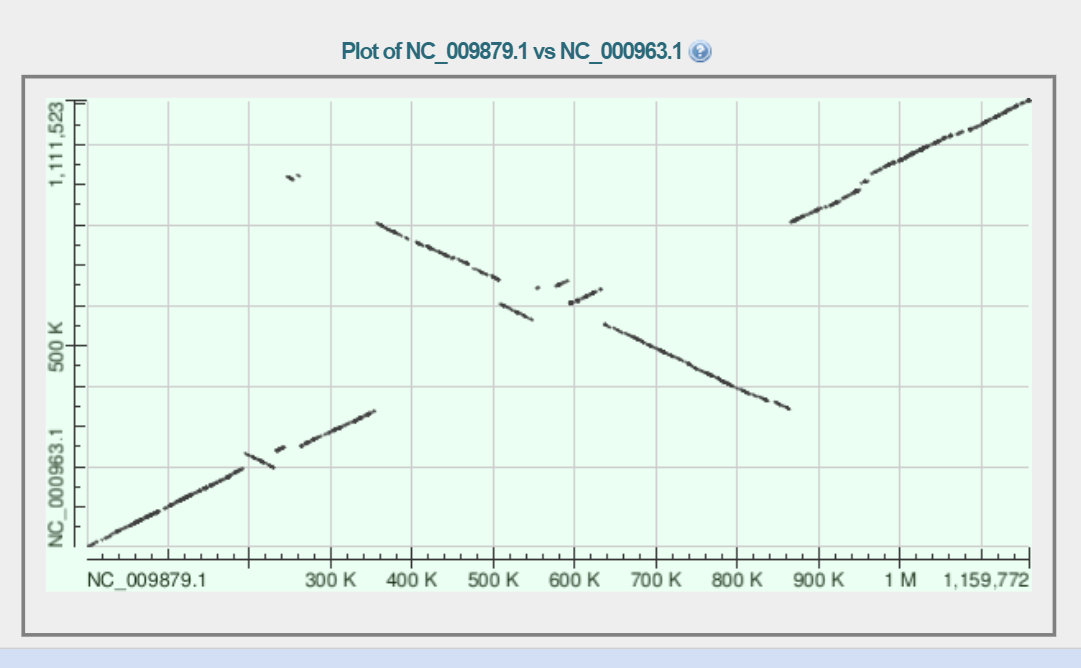 |
Карта локального сходства была получена с помощью MegaBlast с параметрами по умолчанию.
В геноме NC_000963.1 было такое эволюционное событие, как инверсия. В центральной области
сначала идет инверсия, потом короткий участок, где не произошло инверсии, но кажется произошла
дупликация, а потом снова большой инвертированный участок. А так же в начале произошла маленькая инверсия.
© Нестеренко Екатерина 2018