PSI-BLAST
Для выполнения заданий данного практикума была взята аминокислотная последовательность белка аспартатаминотрансферазы из организма бактерии Aquifex aeolicus VF5 (идентификатор в базе данных RefSeq NP_214350). Данная последовательность доступна в формате fasta.
Задание 1.
В данном задании требовалось составить семейство гомологов для последовательности моего белка с помощью программы PSI-BLAST. Для этого я выполнила поиск гомологов последовательности моего белка в данной программе, указав банк UniProtKB/Swiss-Prot, по которому будет производиться поиск, и увеличив количество выводимых на экран находок до 20000, чтобы получать все необходимые последовательности.
Ссылка на проект в JalView.
При повторных запусках программы по уже найденным гомологам обнаруживаются новые последовательности гомологов. Требовалось провести такое количество итераций, чтобы список находок выше порога совпадал со списком последовательностей, поданных на вход. Для каждой последующей итерации программа отбирает находки с E-value < 0.005 (вообще для каждой итерации программа отдельно выдает находки выше и ниже порога - E-value < 0.005).
В процессе выполнения задания я заметила интересную вещь: программа недостаточно строго отсекает находки по порогу, то есть часть находок с E-value < 0.005 оказывается в блоке находок выше порога, часть - ниже. Что любопытно (но вполне очевидно), количество находок в последующих итерациях зависит от количества последовательностей, поданных на вход. Так, в первой итерации получена всего одна находка с E-value = 0.005. Если исключить эту находку из подачи на вход для второй итерации, то будет получено 1847 находок, а если оставить ее, что 1832. Но так как на некоторых итерациях достаточно большое количество находок с E-value = 0.005 оказываются выше порога, то исключать их из списка подаваемых на вход не представляется возможным.
В Таблице 1 представлена информация о каждой итерации (Условия поиска): количество находок лучше порога, появились ли новые находки на данной итерации, ID, score и E-value лучшей и худшей находки (выше порога).
Таблица 1. Сравнение итераций программы PSI-BLAST.
| Номер итерации | Количество находок лучше порога | Появляются ли новые находки | ID лучшей находки выше порога | Score лучшей находки выше порога | E-value лучшей находки выше порога | ID худшей находки выше порога | Score худшей находки выше порога | E-value худшей находки выше порога |
| 1 | 767 | - | O67781.1 | 791 | 0.0 | P74861.3 | 42.4 | 0.005 |
| 2 | 1832 | Да | O67781.1 | 562 | 0.0 | C1CRE4.1 | 42.5 | 0.005 |
| 3 | 3767 | Да | O67781.1 | 443 | 2e-152 | P18486.2 | 42.5 | 0.005 |
| 4 | 4080 | Да | O67781.1 | 371 | 2e-124 | A1CHL0.1 | 42.7 | 0.005 |
| 5 | 4150 | Да | O67781.1 | 330 | 2e-108 | D0ZLR3.1 | 42.3 | 0.005 |
На пятой итерации я заметила, что среди находок есть белки, относящиеся совсем к другим семействам, например, 5-aminolevulinate synthase или Glutamate-1-semialdehyde 2,1-aminomutase. Оказалось, что уже на первой итерации среди находок появляются представители другого семейства 1-aminocyclopropane-1-carboxylate synthase, причем они имеют хороший E-value, и после них еще идут находки семейства аминотрансфераз, что заставляет задуматься о том, можно ли отсечь ненужные находки просто изменением E-value в условиях поиска или в установлении порога PSI-BLAST.
Однако я заметила, что большую часть находок после семейства синтаз составляют Histidinol-phosphate aminotransferase, о гомологичности которых я сомневалась еще в прошлом практикуме, поэтому я решила изменить порог отбора находок для подачи на вход PSI-BLAST и сделала его равным 1e-30, что позволило бы исключить находки 1-aminocyclopropane-1-carboxylate synthase ( Условия поиска). Но при таких параметрах ненужные находки прибавлялись на второй итерации, так как относительно других последовательностей, поданных на вход, их E-value значительно выше.
Тогда я еще уменьшила значение порога, приняв его равным 1e-65 (что позволяет исключать ненужные находки на 2 итерации). И это сработало! Данный порог позволил исключить добавление последовательностей других семейств (только на 3 итерации были получены две находки из семейства лиаз, но я исключила их из подачи на 4 итерацию, поэтому новые лигазы на 4 итерации не добавились).
Таким образом, при поиске с установленным порогом E-value < 1е-65 (Условия поиска) стабилизация находок (отсутствие новых) произошла уже на 4 итерации. Было найдено всего 90 находок, но, как уже говорилось выше, 2 из них - это лиазы, а еще 1 я исключила из-за величины E-value (причины см. ниже) поэтому для выполнения дальнейших заданий я выбрала 87 последовательностей (последовательности доступны в формате fasta). В Таблице 2 представлена информация об итерациях и и характеристиках находок.
О том, что выбранные последовательности составляют одно семество, можно судить по разнице между E-value худшей находки выше порога и лучшей - ниже. Эта разница при последних условиях поиска составляет 10 порядков. Но нетрудно заметить, что последняя находка с E-value = 6e-73 принадлежит возможной аспартатаминотрансферазе, то есть это недостоверный член данного семейства белков. К тому же, если исключить эту последовательность, то разница между E-value находок выше и ниже порога будет составлять 30 порядков (см. Рис. 1). Поэтому для дальнейшего рассмотрения данная последовательность не будет использована.
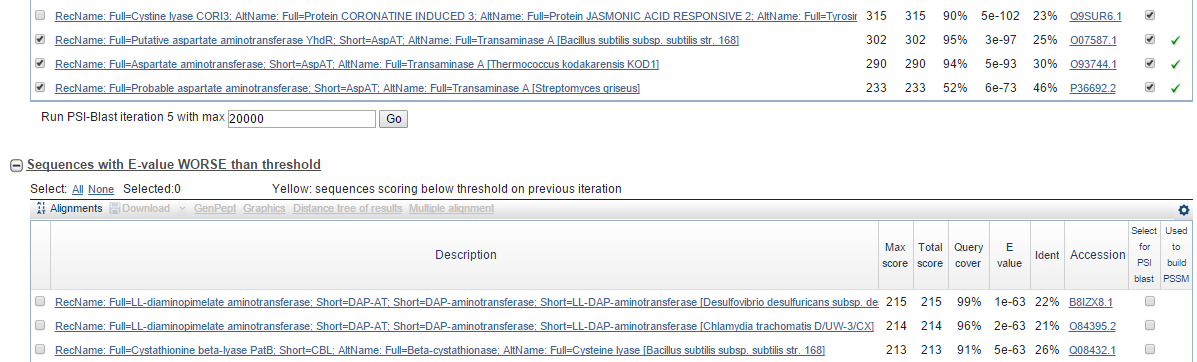
Таблица 2. Сравнение итераций программы PSI-BLAST (E-value < 1e-65).
| Номер итерации | Количество находок лучше порога | Появляются ли новые находки | ID лучшей находки выше порога | Score лучшей находки выше порога | E-value лучшей находки выше порога | ID худшей находки выше порога | Score худшей находки выше порога | E-value худшей находки выше порога |
| 1 | 32 | - | O67781.1 | 791 | 0.0 | Q9HUI9.1 | 222 | 2e-66 |
| 2 | 74 | Да | Q02635.1 | 594 | 0.0 | Q9LVY1.1 | 222 | 2e-66 |
| 3 | 90 | Да | O67781.1 | 528 | 0.0 | Q9SUR6.1 | 226 | 8е-68 |
| 4 | 90 | Нет | O67781.1 | 510 | 7e-179 | Q9SUR6.1 | 233 | 6е-73 |
Задание 3.
В данном задании требовалось выполнить множественное выравнивание отобранных последовательностей с помощью команды muscle на сервере kodomo.
Для этого я использовала команду muscle -in family.fasta -out all_muscle.fasta
С результатом множественного выравнивания можно ознакомиться на Рис. 2 и в прикрепленном проекте JalView (окно all_muscle), а также в формате fasta.

Задание 4.
В данном задании было необходимо построить множественное выравнивание программой muscle уже не всех белков семейства, а специально отобранных seed - 10-20 последовательностей данного семейства, которые должны удовлетворять нескольким критериям:
- они должны быть гомологичны почти по всей длине (coverage = 70-90%)
- гомологии должны быть достоверными (E-value < 1e-3 в результатах поиска)
- в выборке не должно быть "почти идентичных" белков - Identity должна быть на уровне 50-60%
Так как первым двум критериям удовлетворяют все имеющиеся последовательности, то для выбора последовательностей seed я воспользовалась встроенной опцией JalView: Edit -> Remove redundancy. Данная опция оставляет последовательности, сходные меньше, чем на указанное значение порога - процент идентичности последовательностей. Чтобы выбрать необходимое количество последовательностей, я указала порог в 65% идентичности. В результате, я получила 11 последовательностей.
С результатом выбора Seed с помощью опции Remove redundancy и множественным выравниванием этих последовательностей программой muscle можно ознакомиться на Рис. 3 и в окне проекта JalView seed_muscle_remove, а также в формате fasta.

Также я попробовала выполнить операцию по выделению Seed самостоятельно (Seed в формате fasta): в результате 4 итерации PSI-BLAST я выбрала 11 последовательностей с Identity 44%-53% (требуется 50-60%, но выше 53% не было получено). Затем я выполнила множественное выравнивание этих последовательностей по той же схеме, что в задании 2.
Результат эмпирического выбора Seed и его множественного выравнивания программой muscle можно наблюдать на Рис. 4 и в окне JalView seed_muscle_mine (выравнивание в формате fasta).

Выравнивание Seed, выбранного мной лично, мне нравится больше. В дальнейшем буду работать с ним.
Задание 5.
В данном задании было необходимо выполнить множественное выравнивание последовательностей Seed с помощью программы mafft на сервере kodomo.
Для этого я использовала команду mafft seed.fasta > seed_mafft.fasta
С результатом работы данной программы можно ознакомиться на Рис. 5, в проекте JalView в окне seed_mafft, а также в формате fasta.

Задание 6.
В данном задании требуется сравнить полученные с помощью программ muscle и mafft множественные выравнивания Seed семейства гомологов белка аспартатаминотрансферазы.
Для этого я воспользовалась командой muscle -profile -in1 seed_mafft.fasta -in2 seed_muscle_mine.fasta -out both.fasta
С результатом работы программы можно ознакомиться на Рис. 6, в проекте JalView в окне mafft_muscle, а также в формате fasta.
Чтобы сравнить выравнивания, выполненные с помощью программ mafft и muscle (далее просто mafft и muscle соответственно, дабы избежать тавтологии), в их выравнивании друг относительно друга я создала дополнительную строку, в которой я разметила символом I все колонки, совпадающие у muscle и mafft, а символом G - колонки, в которым различие состоит в основном из-за того, что разные программы в разных местах добавляют гэпы, то есть непосредственно от особенностей работы программ muscle и mafft. В неотмеченных колонках различие наблюдается из-за несовпадения аминокислот.
Надо сказать, что это было весьма непростой задачей, так как в каждом из выравниваний последовательности были расположены по-разному.
На Рис. 6 можно видеть вышеупомянутое выравнивание (в верхней части - mafft, в нижней - muscle) и разметку. Выравнивание (общее) получилось очень "хорошее", на Рис. 6 можно наблюдать всего 4 небольших участка различий: 8-11, 25-43, 296-306, 345-362. На Рис. 7 и Рис.8 изображены два из этих участков, 2 и 4, так как там присутствуют и аминокислотные различия, и различия из-за гэпов.

На основе полученного выравнивания крайне трудно провести качественный анализ выравниваний и сделать наполненное смыслом заключение. Даже при подробном подсчете гэпов не удалось выявить, какая же программа ставит их "охотнее", на мой взгляд, их число примерно одинаково с каждой из сторон.