Поиск и описание полиморфизмов у пациента
Данное задание выполнялось для чтений экзома пациента, которые нужно картировать на участок определенной хромосомы человека сборки генома hg19 (в моем случае это хромосома 14 - fasta-файл), которые находятся в файле с одноконцевыми чтениями chr14.fastq).
Часть I: подготовка чтений.
- Анализ качества чтений.
Для начала было необходимо проверить качество полученных нами чтений с помощью программы FastQC.
На Рис. 1а изображена команда, с помощью которой была запущена эта программа.
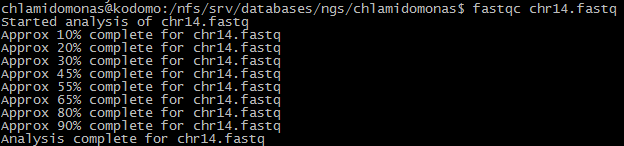
Рис. 1a. Команда, запускающая программу FastQC через Putty.
Результатом работы этой программы является архив chr14_fastqc.zip и файл в формате html, который визуализует разную информацию о чтениях.
На Рис. 1b представлен фрагмент выдачи программы FastQC: картинка "Per base quality", на которой показано качество определения основания в каждой позиции рида длиной 97 п.о. Для визуализации информации используются следующие элементы (соответствующие статистическим характеристикам):- среднее качество (синяя линия)
- медиана - такое значение качества, что половина ридов по данной позиции имеют качество не выше указанного (центральные красные линии)
- интерквартильный размах - разница между верхней и нижней квартилями - такой диапазон значений качества, что 25% чтений имеют значение качества по данной позиции не выше нижней границы, а 75% чтений - не выше верхней (желтый прямоугольник).
- размах между 10-й и 90-й процентилями - 10% чтений имеют значение качества не выше нижней границы, 90% - не выше верхней границы (черные "усы").
Поле графика разделено на полосы разных цветов (зеленая, желтая, красная), попадание вышеупомянутых элементов в которые позволяет судить о качестве чтений (основано на очевидном факте: чем выше значение качества, тем лучше).
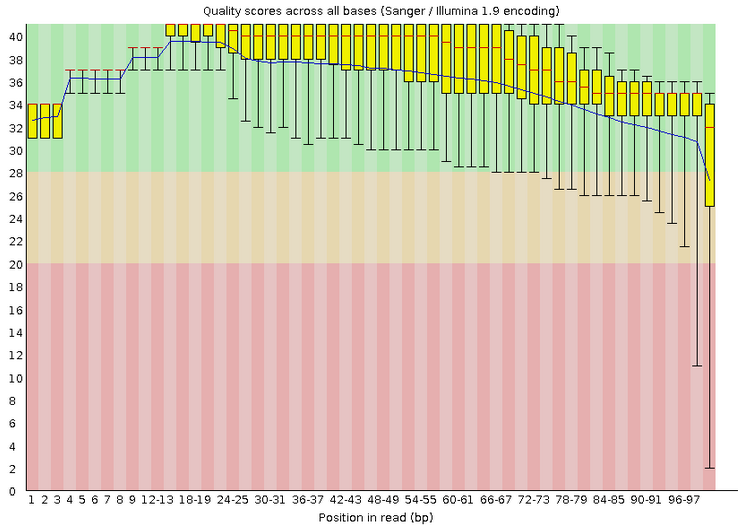
Рис. 1b. Картинка "Per base quality" (фрагмент выдачи программы FastQC), демонстрирующая качество чтений до чистки. По горизонтали: конкретная позиция (или участок) в ридах (1-100), по вертикали: качество определения нуклеотида в позиции.
В данном случае хорошо демонстрируется типичная ситуация: конец ридов прочитан гораздо хуже их основной части - уже с 73 позиции "усы" находятся в желтой области, а в двух последних позициях они находятся в красной области. Это объясняется тем, что к некоторой итерации при секвенировании происходит накопление ошибок (именно поэтому риды не могут быть очень длинными).
Также может быть интересным поле "Basic Statistics" (Рис. 1с), в котором содержится информация о количестве, длине, GC-составе чтений и виде секвенирования. В частности, мы узнаем:- конкретную версию секвенатора Illumina (Illumina 1.9)
- общее число чтений (8696)
- диапазон длины ридов (30-100)
Рис. 1c. Таблица "Basic Statistics" (фрагмент выдачи программы FastQC до чистки). Важная информация о чтениях и виде секвенирования выделена красными рамками.
- Очистка чтений
Далее было нужно очистить чтения с помощью программы Trimmomatic. Известно, что адаптеры в ридах уже удалены.
Я запускала данную программу через сервер kodomo с опциями TRAILING:20 (отрезает с конца каждого чтения нуклеотиды с качеством ниже 20) и MINLEN:50 (удаляет риды короче 50 нуклеотидов) (команда на Рис. 2a). P.S. С выбором опций помогло определиться руководство пользователя.
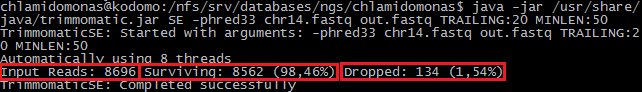
Рис. 2a. Команда, запускающая программу Trimmomatic через Putty. Красными рамками выделены количество ридов до и после чистки и число удаленных чтений.
Таким образом, после чистки осталось 8562 (98,46%) ридов, то есть было удалено 134 (1,54%) чтений.
Затем я снова запустила программу FastQC, чтобы посмотреть, какие риды были удалены при чистке (команда на Рис. 2b).
Рис. 2b. Команда, запускающая программу FastQC через Putty (после чистки).
На Рис. 2с можно увидеть картинку "Per base quality" из выдачи программы FastQC, запущенной после чистки. Хорошо видно, откаких ридов избавился Trimmomatic: исчезли чтения, у которых на концах были нуклеотиды плохого качества. Теперь нет позиций, значения качества которых находятся в красной области, и только последняя позиция расположена в желтой, то есть цель отбора более качественных чтений достигнута.
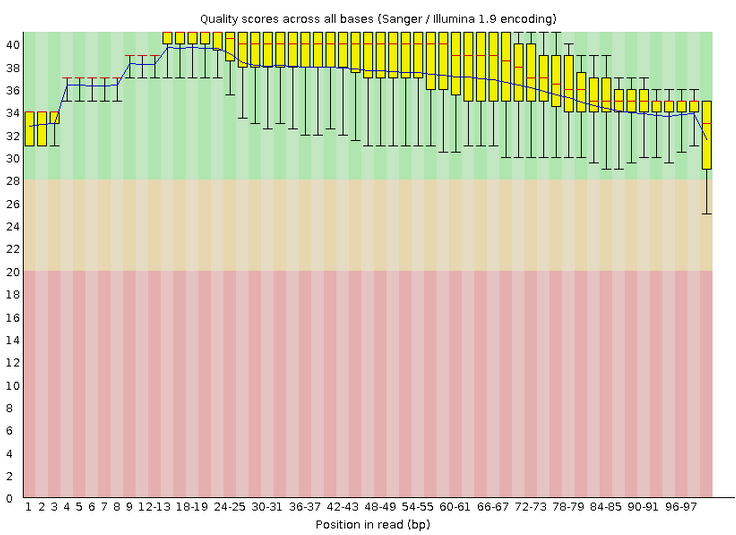
Рис. 2c. Картинка "Per base quality" (фрагмент выдачи программы FastQC), демонстрирующая качество чтений после чистки. По горизонтали: конкретная позиция (или участок) в ридах (1-100), по вертикали: качество определения нуклеотида в позиции.
Еще один результат работы Trimmomatic заметен в таблице "Basic Statistics" (Рис. 2d): были удалены чтения длиной меньше 50 нуклеотидов, и теперь минимальная длины рида равна 50.
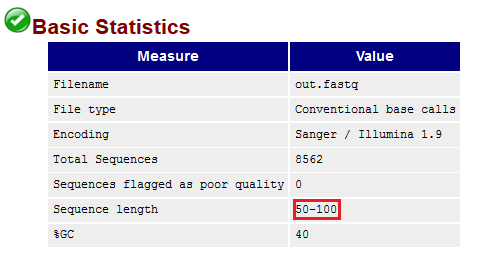
Рис. 2d. Таблица "Basic Statistics" (фрагмент выдачи программы FastQC после чистки). Длина чтений выделена красными рамками.
Часть II: картирование чтений.
- Картирование чтений.
С помощью программы BWA надо было откартировать очищенные чтения. Разобраться с запуском команд мне помогло руководство.
Cначала я проиндексировала референсную последовательность хромосомы 14 (команда на Рис. 3а).
Рис. 3a. Команда bwa index, индексирующая референсную последовательность (программа BWA).
Затем я построила выравнивание прочтений и референса в формате .sam (команда на Рис. 3b).

Рис. 3b. Команда bwa mem, строящая выравнивание прочтений и референсной последовательности (программа BWA).
- Анализ выравнивания.
Данный блок задач был осуществлен с помощью пакета samtools (ссылка на руководство.
Я перевела выравнивание чтений с референсом в бинарный формат .bam (команда на Рис. 4а), запустив команду samtools view с опциями -b (выходной файл бинарный) и -о (указание имени выходного файла).

Рис. 4a. Команда samtools view (пакет samtools).
Затем было нужно отсортировать выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения (команда на Рис. 4b). Это было сделано с помощью команды samtools sort с опцией -T с последующим указанием файла, куда складываются временные файлы.

Рис. 4b. Команда samtools sort (пакет samtools).
Далее я проиндексировала отсортированный .bam файл (команда на Рис. 4с).

Рис. 4c. Команда samtools index (пакет samtools).
И наконец я получила информацию о том, сколько чтений откартировалось на геном (команда на Рис. 4d).

Рис. 4d. Команда samtools idxstats (пакет samtools). Красной рамкой выделена информация о картировании.
Программа выдает информацию о картировании в таком формате: название последовательности, длина последовательности, число картировавшихся ридов, число некартировавшихся ридов. Таким образом, в моем случае картировалось 8563 рида (что странно, так как всего после чистки осталось 8562 чтения), и не было некартировавшихся чтений.
Часть III: анализ SNP.
- Поиск SNP и инделей.
Я воспользовалась командой samtools mpileup с опциями -uf (определяют формат входных и выходных файлов) для получения файла .bcf с полиморфизмами. Отличия между референсом и чтениями были определены с помощью команды bcftools call с опциями -cv пакета bcftools, результатом работы которой является файл в формате .vcf. Команды указаны на Рис. 5а (красное подчеркивание).

Рис. 5a. Команды samtools mpileup (пакет samtools) и bcftools call с опциями -cv пакета bcftools.
Всего было найдено 87 полиморфизмов: 3 вставки, 2 делеции, 82 замены. Из выдачи работы программы я выбрала три полиморфизма и описала их в Таблице 1.
Таблица 1. Описание 3 полиморфизмов.

У первого полимеорфизма "плохие" покрытие и качество, а у остальных двух качество хорошее, а покрытие ниже среднего и среднее, соответственно (максимальное покрытие 70).
- Аннотация SNP.
Далее нужно было проаннотировать только полученные snp помощью программы annovar по следующим базам данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Программа annovar установлена на kodomo: /nfs/srv/databases/annovar.
Программа умеет работать с файлами в формате avinput. Получить этот файл из файла .vcf можно с помощью скрипта convert2annovar.pl (команда на Рис. 6). Для аннотации файла с snp с помощью предложенных баз данных использовался скрипт annotate_variation.pl.
Рис. 6. Команды perl convert2annovar.pl (получение файла в формате avinput) и perl annotate_variation.pl - информация о том, какие замены имеют rs.
В качестве примера на Рис. 6 приведена команда, которая позволяет узнать, какие из найденных замен имеют rs (идентификатор в базе данных однонуклеотидных полиморфизмов - то есть уже изучены или хотя бы известны). Команды, с помощью которых можно проаннотировать полиморфизмы по указанным базам данных, приведены в Таблице 3.
Необходимые для анализа результаты аннотаций по этим базам данных представлены в файле Excel.
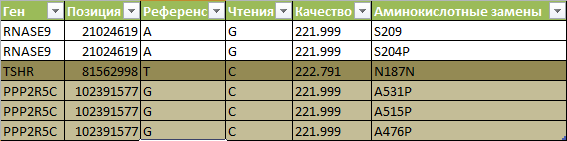
База данных refseq выделила 29 het замен и 53 hom, из них 3 замены попали в экзоны, одна - в UTR3 (3'-нетранслируемую область), одна в сплайсинг-вариант, а остальные 77 в интроны. Замены затронули гены RNASE9, PPP2R5C, TSHR, по одной замене на экзоны каждого гена. TSHR - ген рецептора тироид-стимулирующего гормона, RNASE9 - ген рибонуклеазы 9 из семейства РНКаз А, PPP2R5C - ген регуляторной субъединицы гамма белковой фосфатазы 2. Из 3 произошедших замен в экзонах замена в гене TSHR является синонимичной (не привела к изменению аминокислоты), а замены в двух оставшихся генах были несинонимичные (привели к замене аминокислоты).
В Таблице 2 представлена информация о нуклеотидных и аминокислотных заменах в этих генах.
Таблица 2. Описание нуклеотидных и аминокислотных замен.
Аннотация по базе dbsnp показала, что 76 замен имеют rs (лист dbs.hg19_snp138_dropped в файле Excel), а 6 замен не имеют (лист dbs.hg19_snp138_filtered в файле Excel).
Частоты замен были определены по базе 1000 genomes (лист 1000g.hg19_dropped в файле Excel). Двумя самыми часто распространенными заменами в данной хромосоме является замена тимина на аденин или цитозин в TSHR - гене рецептора тироид-стимулирующего гормона, обе происходят в интронах (Рис. 9). Наиболее распространенная частота замен - от 0.3 до 0.5.
Клиническая аннотация была получена из базы GWAS (лист gwas в файле Excel): замена в экзоне гена RNASE9 связана с раком простаты, в интроне гена TSHR - с болезнью Грейвса, а в интроне гена PPP2R5C - с аутизмом.
В качестве обобщения результатов аннотирования привожу сводную таблицу.
В Таблице 3 перечислены все использованные команды.