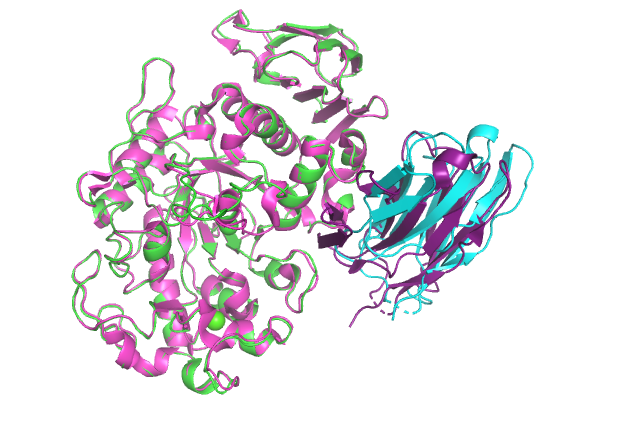
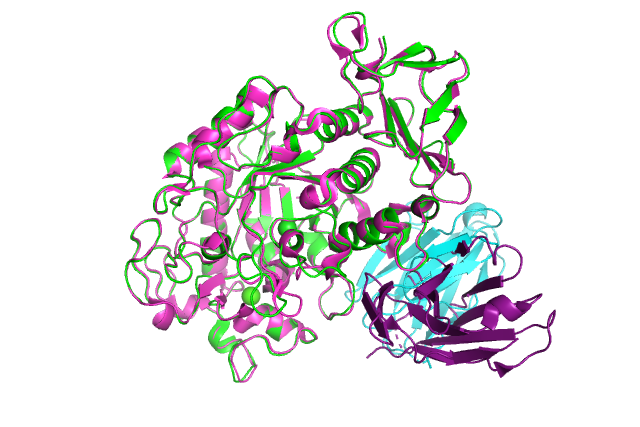
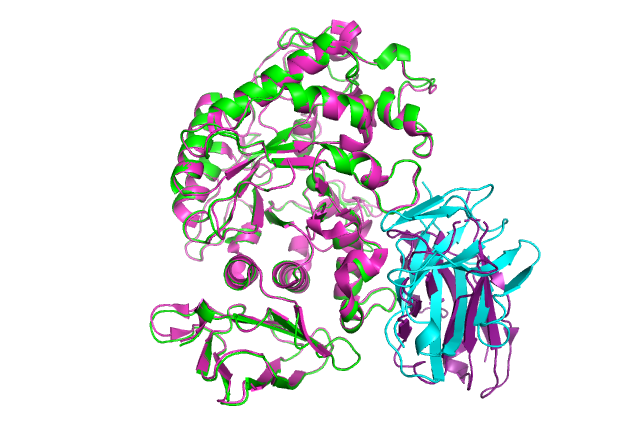
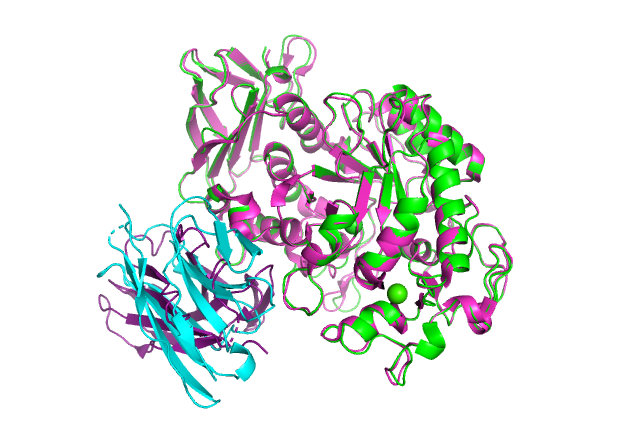
:-) G R O M A C S (-:
Good ROcking Metal Altar for Chronical Sinners
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) pdb2gmx (-:
Option Filename Type Description
------------------------------------------------------------
-f amylase.pdb Input Structure file: gro g96 pdb tpr etc.
-o amylase_h.pdb Output Structure file: gro g96 pdb etc.
-p topol.top Output Topology file
-i posre.itp Output Include file for topology
-n clean.ndx Output, Opt. Index file
-q clean.pdb Output, Opt. Structure file: gro g96 pdb etc.
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-chainsep enum id_or_ter Condition in PDB files when a new chain should
be started (adding termini): id_or_ter,
id_and_ter, ter, id or interactive
-merge enum no Merge multiple chains into a single
[moleculetype]: no, all or interactive
-ff string gromos53a6 Force field, interactive by default. Use -h
for information.
-water enum none Water model to use: select, none, spc, spce,
tip3p, tip4p or tip5p
-[no]inter bool no Set the next 8 options to interactive
-[no]ss bool no Interactive SS bridge selection
-[no]ter bool no Interactive termini selection, instead of charged
(default)
-[no]lys bool no Interactive lysine selection, instead of charged
-[no]arg bool no Interactive arginine selection, instead of charged
-[no]asp bool no Interactive aspartic acid selection, instead of
charged
-[no]glu bool no Interactive glutamic acid selection, instead of
charged
-[no]gln bool no Interactive glutamine selection, instead of
neutral
-[no]his bool no Interactive histidine selection, instead of
checking H-bonds
-angle real 135 Minimum hydrogen-donor-acceptor angle for a
H-bond (degrees)
-dist real 0.3 Maximum donor-acceptor distance for a H-bond (nm)
-[no]una bool no Select aromatic rings with united CH atoms on
phenylalanine, tryptophane and tyrosine
-[no]ignh bool yes Ignore hydrogen atoms that are in the coordinate
file
-[no]missing bool no Continue when atoms are missing, dangerous
-[no]v bool no Be slightly more verbose in messages
-posrefc real 1000 Force constant for position restraints
-vsite enum none Convert atoms to virtual sites: none, hydrogens
or aromatics
-[no]heavyh bool no Make hydrogen atoms heavy
-[no]deuterate bool no Change the mass of hydrogens to 2 amu
-[no]chargegrp bool yes Use charge groups in the .rtp file
-[no]cmap bool yes Use cmap torsions (if enabled in the .rtp file)
-[no]renum bool no Renumber the residues consecutively in the output
-[no]rtpres bool no Use .rtp entry names as residue names
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.r2b
All occupancies are one
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/atomtypes.atp
Atomtype 1
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.rtp
Using default: not generating all possible dihedrals
Using default: excluding 3 bonded neighbors
Using default: generating 1,4 H--H interactions
Using default: removing impropers on same bond as a proper
Residue 108
Sorting it all out...
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.hdb
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.n.tdb
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.c.tdb
Back Off! I just backed up topol.top to ./#topol.top.2#
Analysing hydrogen-bonding network for automated assignment of histidine
protonation. 761 donors and 727 acceptors were found.
There are 1112 hydrogen bonds
Will use HISE for residue 15
Will use HISE for residue 101
Will use HISD for residue 201
Will use HISD for residue 215
Will use HISE for residue 299
Will use HISE for residue 305
Will use HISE for residue 331
Will use HISE for residue 386
Will use HISE for residue 491
8 out of 8 lines of specbond.dat converted successfully
Special Atom Distance matrix:
HIS15 CYS28 CYS70 MET82 CYS86 HIS101 MET102
NE2108 SG226 SG568 SD667 SG699 NE2817 SD824
CYS28 SG226 1.477
CYS70 SG568 2.005 2.128
MET82 SD667 1.264 0.664 1.475
CYS86 SG699 1.513 0.203 1.981 0.534
HIS101 NE2817 1.466 2.637 1.962 2.164 2.578
MET102 SD824 1.986 2.772 1.528 2.188 2.655 0.870
CYS103 SG831 2.123 3.077 1.577 2.469 2.979 1.037 0.737
CYS115 SG895 2.157 2.309 0.203 1.653 2.156 1.985 1.471
CYS119 SG923 2.402 3.163 1.363 2.518 3.043 1.469 0.923
CYS141 SG1096 2.888 4.102 3.204 3.640 4.040 1.497 1.749
CYS160 SG1242 2.772 3.997 3.205 3.558 3.941 1.420 1.753
MET178 SD1384 2.656 2.994 1.850 2.453 2.834 1.782 1.043
HIS201 NE21563 1.740 3.049 2.737 2.701 3.028 0.792 1.580
MET202 SD1570 1.804 2.720 2.352 2.331 2.649 0.825 1.111
HIS215 NE21676 2.642 2.416 2.317 2.122 2.258 2.423 2.034
MET274 SD2144 2.653 3.240 4.641 3.546 3.408 3.660 4.424
MET287 SD2255 1.849 2.658 3.493 2.682 2.706 2.091 2.739
HIS299 NE22352 0.536 1.992 2.086 1.692 2.008 1.065 1.743
HIS305 NE22402 1.473 2.809 3.217 2.711 2.898 2.036 2.850
MET328 SD2572 1.412 2.027 3.367 2.252 2.160 2.491 3.157
HIS331 NE22596 2.492 2.925 4.402 3.239 3.060 3.331 4.014
MET339 SD2662 0.507 1.636 2.455 1.598 1.730 1.845 2.456
CYS378 SG2970 1.529 1.356 2.560 1.545 1.497 2.917 3.271
CYS384 SG3017 1.340 1.315 2.498 1.476 1.456 2.742 3.127
HIS386 NE23036 1.257 0.955 2.517 1.270 1.126 2.699 3.069
MET394 SD3115 2.231 2.212 4.016 2.685 2.399 3.533 4.122
CYS450 SG3564 3.335 2.796 4.811 3.404 2.991 4.742 5.228
CYS462 SG3645 3.478 2.975 4.970 3.576 3.172 4.888 5.389
HIS491 NE23860 2.628 2.179 4.197 2.766 2.367 3.964 4.451
CYS103 CYS115 CYS119 CYS141 CYS160 MET178 HIS201
SG831 SG895 SG923 SG1096 SG1242 SD1384 NE21563
CYS115 SG895 1.515
CYS119 SG923 0.491 1.251
CYS141 SG1096 1.755 3.157 2.151
CYS160 SG1242 1.802 3.169 2.218 0.203
MET178 SD1384 1.689 1.750 1.635 2.374 2.384
HIS201 NE21563 1.711 2.771 2.183 1.277 1.117 2.360
MET202 SD1570 1.644 2.362 1.991 1.560 1.442 1.536 0.963
HIS215 NE21676 2.738 2.313 2.748 3.305 3.242 1.362 2.862
MET274 SD2144 4.540 4.803 4.922 4.537 4.358 5.048 3.318
MET287 SD2255 3.121 3.590 3.516 2.799 2.607 3.134 1.690
HIS299 NE22352 1.799 2.205 2.156 2.412 2.297 2.554 1.272
HIS305 NE22402 2.743 3.343 3.150 2.914 2.779 3.729 1.791
MET328 SD2572 3.385 3.522 3.729 3.596 3.426 3.691 2.323
HIS331 NE22596 4.314 4.545 4.689 4.169 3.976 4.438 2.974
MET339 SD2662 2.537 2.616 2.840 3.171 3.041 3.151 1.957
CYS378 SG2970 3.289 2.761 3.425 4.358 4.256 3.837 3.236
CYS384 SG3017 3.151 2.697 3.310 4.175 4.070 3.712 3.041
HIS386 NE23036 3.203 2.714 3.369 4.145 4.028 3.557 2.979
MET394 SD3115 4.341 4.196 4.630 4.718 4.551 4.578 3.443
CYS450 SG3564 5.444 5.005 5.662 6.024 5.865 5.594 4.755
CYS462 SG3645 5.589 5.165 5.811 6.160 6.000 5.774 4.887
HIS491 NE23860 4.730 4.381 4.970 5.214 5.051 4.783 3.955
MET202 HIS215 MET274 MET287 HIS299 HIS305 MET328
SD1570 NE21676 SD2144 SD2255 NE22352 NE22402 SD2572
HIS215 NE21676 1.936
MET274 SD2144 3.747 4.709
MET287 SD2255 1.755 2.839 2.285
HIS299 NE22352 1.539 2.768 2.768 1.792
HIS305 NE22402 2.481 3.921 2.025 2.016 1.238
MET328 SD2572 2.515 3.322 1.406 1.306 1.624 1.553
HIS331 NE22596 3.161 3.913 1.193 1.475 2.632 2.349 1.118
MET339 SD2662 2.180 3.077 2.197 1.837 0.796 1.173 1.088
CYS378 SG2970 3.300 3.606 2.788 3.050 1.970 2.271 1.998
CYS384 SG3017 3.122 3.499 2.664 2.862 1.779 2.088 1.827
HIS386 NE23036 2.958 3.195 2.565 2.598 1.765 2.188 1.590
MET394 SD3115 3.569 4.001 1.309 2.309 2.583 2.394 1.131
CYS450 SG3564 4.794 4.856 2.361 3.623 3.769 3.623 2.482
CYS462 SG3645 4.954 5.052 2.384 3.762 3.901 3.703 2.607
HIS491 NE23860 3.938 4.036 1.953 2.750 3.053 3.044 1.699
HIS331 MET339 CYS378 CYS384 HIS386 MET394 CYS450
NE22596 SD2662 SG2970 SG3017 NE23036 SD3115 SG3564
MET339 SD2662 2.198
CYS378 SG2970 3.007 1.364
CYS384 SG3017 2.860 1.162 0.202
HIS386 NE23036 2.587 1.119 0.563 0.470
MET394 SD3115 1.364 1.855 1.999 1.918 1.664
CYS450 SG3564 2.558 2.995 2.514 2.545 2.340 1.371
CYS462 SG3645 2.655 3.121 2.629 2.663 2.483 1.486 0.202
HIS491 NE23860 1.824 2.332 2.180 2.149 1.829 0.710 0.917
CYS462
SG3645
HIS491 NE23860 1.091
Linking CYS-28 SG-226 and CYS-86 SG-699...
Linking CYS-70 SG-568 and CYS-115 SG-895...
Linking CYS-141 SG-1096 and CYS-160 SG-1242...
Linking CYS-378 SG-2970 and CYS-384 SG-3017...
Linking CYS-450 SG-3564 and CYS-462 SG-3645...
Making bonds...
Number of bonds was 5207, now 5202
Generating angles, dihedrals and pairs...
Before cleaning: 8104 pairs
Before cleaning: 10145 dihedrals
Making cmap torsions...There are 2623 dihedrals, 2788 impropers, 7653 angles
8104 pairs, 5202 bonds and 0 virtual sites
Total mass 55217.920 a.m.u.
Total charge -6.000 e
Writing topology
Back Off! I just backed up posre.itp to ./#posre.itp.2#
Writing coordinate file...
Back Off! I just backed up amylase_h.pdb to ./#amylase_h.pdb.1#
:-) G R O M A C S (-:
Great Red Oystrich Makes All Chemists Sane
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) pdb2gmx (-:
Option Filename Type Description
------------------------------------------------------------
-f camelid.pdb Input Structure file: gro g96 pdb tpr etc.
-o camelid_h.pdb Output Structure file: gro g96 pdb etc.
-p topol.top Output Topology file
-i posre.itp Output Include file for topology
-n clean.ndx Output, Opt. Index file
-q clean.pdb Output, Opt. Structure file: gro g96 pdb etc.
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-chainsep enum id_or_ter Condition in PDB files when a new chain should
be started (adding termini): id_or_ter,
id_and_ter, ter, id or interactive
-merge enum no Merge multiple chains into a single
[moleculetype]: no, all or interactive
-ff string gromos53a6 Force field, interactive by default. Use -h
for information.
-water enum none Water model to use: select, none, spc, spce,
tip3p, tip4p or tip5p
-[no]inter bool no Set the next 8 options to interactive
-[no]ss bool no Interactive SS bridge selection
-[no]ter bool no Interactive termini selection, instead of charged
(default)
-[no]lys bool no Interactive lysine selection, instead of charged
-[no]arg bool no Interactive arginine selection, instead of charged
-[no]asp bool no Interactive aspartic acid selection, instead of
charged
-[no]glu bool no Interactive glutamic acid selection, instead of
charged
-[no]gln bool no Interactive glutamine selection, instead of
neutral
-[no]his bool no Interactive histidine selection, instead of
checking H-bonds
-angle real 135 Minimum hydrogen-donor-acceptor angle for a
H-bond (degrees)
-dist real 0.3 Maximum donor-acceptor distance for a H-bond (nm)
-[no]una bool no Select aromatic rings with united CH atoms on
phenylalanine, tryptophane and tyrosine
-[no]ignh bool yes Ignore hydrogen atoms that are in the coordinate
file
-[no]missing bool no Continue when atoms are missing, dangerous
-[no]v bool no Be slightly more verbose in messages
-posrefc real 1000 Force constant for position restraints
-vsite enum none Convert atoms to virtual sites: none, hydrogens
or aromatics
-[no]heavyh bool no Make hydrogen atoms heavy
-[no]deuterate bool no Change the mass of hydrogens to 2 amu
-[no]chargegrp bool yes Use charge groups in the .rtp file
-[no]cmap bool yes Use cmap torsions (if enabled in the .rtp file)
-[no]renum bool no Renumber the residues consecutively in the output
-[no]rtpres bool no Use .rtp entry names as residue names
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.r2b
All occupancies are one
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/atomtypes.atp
Atomtype 1
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.rtp
Using default: not generating all possible dihedrals
Using default: excluding 3 bonded neighbors
Using default: generating 1,4 H--H interactions
Using default: removing impropers on same bond as a proper
Residue 108
Sorting it all out...
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.hdb
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.n.tdb
Opening force field file /usr/share/gromacs/top/gromos53a6.ff/aminoacids.c.tdb
Back Off! I just backed up topol.top to ./#topol.top.3#
8 out of 8 lines of specbond.dat converted successfully
Special Atom Distance matrix:
CYS22 MET34 CYS45 MET82 CYS92
SG143 SD215 SG306 SD588 SG693
MET34 SD215 0.667
CYS45 SG306 1.465 1.705
MET82 SD588 1.290 1.645 1.477
CYS92 SG693 0.204 0.644 1.283 1.231
CYS100 SG792 1.473 1.646 0.204 1.609 1.284
Linking CYS-22 SG-143 and CYS-92 SG-693...
Linking CYS-45 SG-306 and CYS-100 SG-792...
Making bonds...
Warning: Long Bond (201-203 = 0.338522 nm)
Number of bonds was 1202, now 1197
Generating angles, dihedrals and pairs...
Before cleaning: 1935 pairs
Before cleaning: 2304 dihedrals
Making cmap torsions...There are 628 dihedrals, 603 impropers, 1748 angles
1935 pairs, 1197 bonds and 0 virtual sites
Total mass 12878.351 a.m.u.
Total charge 2.000 e
Writing topology
Back Off! I just backed up posre.itp to ./#posre.itp.3#
Writing coordinate file...
Back Off! I just backed up camelid_h.pdb to ./#camelid_h.pdb.1#