Молекулярная динамика биологических молекул в GROMACS
Моделирование самосборки липидного бислоя
Создадим ячейку из 64 молекул одного липида.
In [1]:
%%bash
genconf -f dppc.gro -o b_64.gro -nbox 4 4 4
:-) G R O M A C S (-:
Gnomes, ROck Monsters And Chili Sauce
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) genconf (-:
Option Filename Type Description
------------------------------------------------------------
-f dppc.gro Input Structure file: gro g96 pdb tpr etc.
-o b_64.gro Output Structure file: gro g96 pdb etc.
-trj traj.xtc Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-nbox vector 4 4 4 Number of boxes
-dist vector 0 0 0 Distance between boxes
-seed int 0 Random generator seed, if 0 generated from the
time
-[no]rot bool no Randomly rotate conformations
-[no]shuffle bool no Random shuffling of molecules
-[no]sort bool no Sort molecules on X coord
-block int 1 Divide the box in blocks on this number of cpus
-nmolat int 3 Number of atoms per molecule, assumed to start
from 0. If you set this wrong, it will screw up
your system!
-maxrot vector 180 180 180 Maximum random rotation
-[no]renumber bool yes Renumber residues
gcq#162: "Confirmed" (Star Trek)
In [3]:
%%bash
editconf -f dppc.gro -o dppc.pdb
Read 50 atoms Volume: 1.5477 nm^3, corresponds to roughly 600 electrons No velocities found
:-) G R O M A C S (-:
Gnomes, ROck Monsters And Chili Sauce
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) editconf (-:
Option Filename Type Description
------------------------------------------------------------
-f dppc.gro Input Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-o dppc.pdb Output, Opt! Structure file: gro g96 pdb etc.
-mead mead.pqr Output, Opt. Coordinate file for MEAD
-bf bfact.dat Input, Opt. Generic data file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-[no]ndef bool no Choose output from default index groups
-bt enum triclinic Box type for -box and -d: triclinic, cubic,
dodecahedron or octahedron
-box vector 0 0 0 Box vector lengths (a,b,c)
-angles vector 90 90 90 Angles between the box vectors (bc,ac,ab)
-d real 0 Distance between the solute and the box
-[no]c bool no Center molecule in box (implied by -box and -d)
-center vector 0 0 0 Coordinates of geometrical center
-aligncenter vector 0 0 0 Center of rotation for alignment
-align vector 0 0 0 Align to target vector
-translate vector 0 0 0 Translation
-rotate vector 0 0 0 Rotation around the X, Y and Z axes in degrees
-[no]princ bool no Orient molecule(s) along their principal axes
-scale vector 1 1 1 Scaling factor
-density real 1000 Density (g/L) of the output box achieved by
scaling
-[no]pbc bool no Remove the periodicity (make molecule whole again)
-resnr int -1 Renumber residues starting from resnr
-[no]grasp bool no Store the charge of the atom in the B-factor
field and the radius of the atom in the occupancy
field
-rvdw real 0.12 Default Van der Waals radius (in nm) if one can
not be found in the database or if no parameters
are present in the topology file
-[no]sig56 bool no Use rmin/2 (minimum in the Van der Waals
potential) rather than sigma/2
-[no]vdwread bool no Read the Van der Waals radii from the file
vdwradii.dat rather than computing the radii
based on the force field
-[no]atom bool no Force B-factor attachment per atom
-[no]legend bool no Make B-factor legend
-label string A Add chain label for all residues
-[no]conect bool no Add CONECT records to a .pdb file when written.
Can only be done when a topology is present
gcq#159: "Way to Go Dude" (Beavis and Butthead)
In [4]:
%%bash
editconf -f b_64.gro -o b_64.pdb
Read 3200 atoms Volume: 99.0529 nm^3, corresponds to roughly 44500 electrons No velocities found
:-) G R O M A C S (-:
Glycine aRginine prOline Methionine Alanine Cystine Serine
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) editconf (-:
Option Filename Type Description
------------------------------------------------------------
-f b_64.gro Input Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-o b_64.pdb Output, Opt! Structure file: gro g96 pdb etc.
-mead mead.pqr Output, Opt. Coordinate file for MEAD
-bf bfact.dat Input, Opt. Generic data file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-[no]ndef bool no Choose output from default index groups
-bt enum triclinic Box type for -box and -d: triclinic, cubic,
dodecahedron or octahedron
-box vector 0 0 0 Box vector lengths (a,b,c)
-angles vector 90 90 90 Angles between the box vectors (bc,ac,ab)
-d real 0 Distance between the solute and the box
-[no]c bool no Center molecule in box (implied by -box and -d)
-center vector 0 0 0 Coordinates of geometrical center
-aligncenter vector 0 0 0 Center of rotation for alignment
-align vector 0 0 0 Align to target vector
-translate vector 0 0 0 Translation
-rotate vector 0 0 0 Rotation around the X, Y and Z axes in degrees
-[no]princ bool no Orient molecule(s) along their principal axes
-scale vector 1 1 1 Scaling factor
-density real 1000 Density (g/L) of the output box achieved by
scaling
-[no]pbc bool no Remove the periodicity (make molecule whole again)
-resnr int -1 Renumber residues starting from resnr
-[no]grasp bool no Store the charge of the atom in the B-factor
field and the radius of the atom in the occupancy
field
-rvdw real 0.12 Default Van der Waals radius (in nm) if one can
not be found in the database or if no parameters
are present in the topology file
-[no]sig56 bool no Use rmin/2 (minimum in the Van der Waals
potential) rather than sigma/2
-[no]vdwread bool no Read the Van der Waals radii from the file
vdwradii.dat rather than computing the radii
based on the force field
-[no]atom bool no Force B-factor attachment per atom
-[no]legend bool no Make B-factor legend
-label string A Add chain label for all residues
-[no]conect bool no Add CONECT records to a .pdb file when written.
Can only be done when a topology is present
gcq#315: "Nobody Never Learnt No-Nothing from No History" (Gogol Bordello)
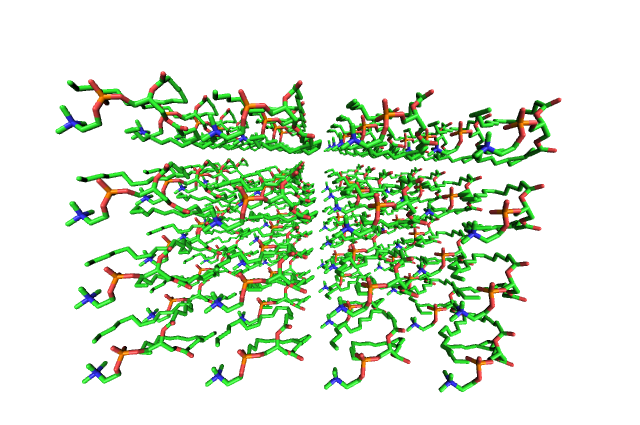
Создадим в ячейке отступ от липидов примерно для 2500 молекул воды.
In [6]:
%%bash
editconf -f b_64.gro -o b_ec -d 0.5
Read 3200 atoms
Volume: 99.0529 nm^3, corresponds to roughly 44500 electrons
No velocities found
system size : 5.260 3.443 4.778 (nm)
center : 2.730 1.822 2.490 (nm)
box vectors : 5.460 3.643 4.979 (nm)
box angles : 90.00 90.00 90.00 (degrees)
box volume : 99.05 (nm^3)
shift : 0.400 0.400 0.399 (nm)
new center : 3.130 2.222 2.889 (nm)
new box vectors : 6.260 4.443 5.778 (nm)
new box angles : 90.00 90.00 90.00 (degrees)
new box volume : 160.70 (nm^3)
WARNING: No boxtype specified - distance condition applied in each dimension.
If the molecule rotates the actual distance will be smaller. You might want
to use a cubic box instead, or why not try a dodecahedron today?
:-) G R O M A C S (-:
Gyas ROwers Mature At Cryogenic Speed
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) editconf (-:
Option Filename Type Description
------------------------------------------------------------
-f b_64.gro Input Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-o b_ec.gro Output, Opt! Structure file: gro g96 pdb etc.
-mead mead.pqr Output, Opt. Coordinate file for MEAD
-bf bfact.dat Input, Opt. Generic data file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-[no]ndef bool no Choose output from default index groups
-bt enum triclinic Box type for -box and -d: triclinic, cubic,
dodecahedron or octahedron
-box vector 0 0 0 Box vector lengths (a,b,c)
-angles vector 90 90 90 Angles between the box vectors (bc,ac,ab)
-d real 0.5 Distance between the solute and the box
-[no]c bool no Center molecule in box (implied by -box and -d)
-center vector 0 0 0 Coordinates of geometrical center
-aligncenter vector 0 0 0 Center of rotation for alignment
-align vector 0 0 0 Align to target vector
-translate vector 0 0 0 Translation
-rotate vector 0 0 0 Rotation around the X, Y and Z axes in degrees
-[no]princ bool no Orient molecule(s) along their principal axes
-scale vector 1 1 1 Scaling factor
-density real 1000 Density (g/L) of the output box achieved by
scaling
-[no]pbc bool no Remove the periodicity (make molecule whole again)
-resnr int -1 Renumber residues starting from resnr
-[no]grasp bool no Store the charge of the atom in the B-factor
field and the radius of the atom in the occupancy
field
-rvdw real 0.12 Default Van der Waals radius (in nm) if one can
not be found in the database or if no parameters
are present in the topology file
-[no]sig56 bool no Use rmin/2 (minimum in the Van der Waals
potential) rather than sigma/2
-[no]vdwread bool no Read the Van der Waals radii from the file
vdwradii.dat rather than computing the radii
based on the force field
-[no]atom bool no Force B-factor attachment per atom
-[no]legend bool no Make B-factor legend
-label string A Add chain label for all residues
-[no]conect bool no Add CONECT records to a .pdb file when written.
Can only be done when a topology is present
gcq#49: "You Could Make More Money As a Butcher" (F. Zappa)
Оптимизируем геометрию системы, чтобы удалить "плохие" контакты молекул.
In [7]:
%%bash
grompp -f em -c b_ec -p b -o b_em -maxwarn 2
mdrun -deffnm b_em -v
:-) G R O M A C S (-:
Good gRace! Old Maple Actually Chews Slate
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) grompp (-:
Analysing residue names:
There are: 64 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Largest charge group radii for Van der Waals: 0.235, 0.235 nm
Largest charge group radii for Coulomb: 0.235, 0.235 nm
This run will generate roughly 466 Mb of data
Option Filename Type Description
------------------------------------------------------------
-f em.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c b_ec.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p b.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o b_em.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]v bool no Be loud and noisy
-time real -1 Take frame at or first after this time.
-[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
-maxwarn int 2 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
-[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
-[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Ignoring obsolete mdp entry 'title'
Ignoring obsolete mdp entry 'cpp'
Generated 1369 of the 2211 non-bonded parameter combinations
Excluding 3 bonded neighbours molecule type 'DPPC'
Number of degrees of freedom in T-Coupling group rest is 9597.00
NOTE 1 [file em.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
WARNING 1 [file em.mdp]:
The sum of the two largest charge group radii (0.469576) is larger than
rlist (0.350000)
There was 1 note
There was 1 warning
gcq#5: "We Can Dance Like Iggy Pop" (Red Hot Chili Peppers)
:-) G R O M A C S (-:
Good gRace! Old Maple Actually Chews Slate
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s b_em.tpr Input Run input file: tpr tpb tpa
-o b_em.trr Output Full precision trajectory: trr trj cpt
-x b_em.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi b_em.cpt Input, Opt. Checkpoint file
-cpo b_em.cpt Output, Opt. Checkpoint file
-c b_em.gro Output Structure file: gro g96 pdb etc.
-e b_em.edr Output Energy file
-g b_em.log Output Log file
-dhdl b_em.xvg Output, Opt. xvgr/xmgr file
-field b_em.xvg Output, Opt. xvgr/xmgr file
-table b_em.xvg Input, Opt. xvgr/xmgr file
-tablep b_em.xvg Input, Opt. xvgr/xmgr file
-tableb b_em.xvg Input, Opt. xvgr/xmgr file
-rerun b_em.xtc Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi b_em.xvg Output, Opt. xvgr/xmgr file
-tpid b_em.xvg Output, Opt. xvgr/xmgr file
-ei b_em.edi Input, Opt. ED sampling input
-eo b_em.edo Output, Opt. ED sampling output
-j b_em.gct Input, Opt. General coupling stuff
-jo b_em.gct Output, Opt. General coupling stuff
-ffout b_em.xvg Output, Opt. xvgr/xmgr file
-devout b_em.xvg Output, Opt. xvgr/xmgr file
-runav b_em.xvg Output, Opt. xvgr/xmgr file
-px b_em.xvg Output, Opt. xvgr/xmgr file
-pf b_em.xvg Output, Opt. xvgr/xmgr file
-mtx b_em.mtx Output, Opt. Hessian matrix
-dn b_em.ndx Output, Opt. Index file
-multidir b_em Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string b_em Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 0 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Getting Loaded...
Reading file b_em.tpr, VERSION 4.5.5 (single precision)
Starting 8 threads
Loaded with Money
Making 3D domain decomposition 2 x 2 x 2
Steepest Descents:
Tolerance (Fmax) = 1.00000e+00
Number of steps = 1000000
Step= 0, Dmax= 2.0e-02 nm, Epot= 4.74007e+05 Fmax= 4.37970e+05, atom= 1842
Step= 1, Dmax= 2.0e-02 nm, Epot= 7.19293e+04 Fmax= 3.57933e+04, atom= 2588
Step= 2, Dmax= 2.4e-02 nm, Epot= 3.56795e+04 Fmax= 9.52568e+03, atom= 2993
Step= 3, Dmax= 2.9e-02 nm, Epot= 3.46459e+04 Fmax= 1.50508e+04, atom= 2992
Step= 4, Dmax= 3.5e-02 nm, Epot= 2.99879e+04 Fmax= 1.37059e+04, atom= 2592
Step= 6, Dmax= 2.1e-02 nm, Epot= 1.30782e+04 Fmax= 4.02791e+03, atom= 1342
Step= 9, Dmax= 6.2e-03 nm, Epot= 1.04672e+04 Fmax= 1.87751e+03, atom= 2392
Step= 10, Dmax= 7.5e-03 nm, Epot= 1.02199e+04 Fmax= 3.76147e+03, atom= 1792
Step= 11, Dmax= 9.0e-03 nm, Epot= 1.01147e+04 Fmax= 4.34365e+03, atom= 2392
Step= 13, Dmax= 5.4e-03 nm, Epot= 7.19423e+03 Fmax= 1.91912e+03, atom= 2765
Step= 15, Dmax= 3.2e-03 nm, Epot= 6.81202e+03 Fmax= 2.03533e+03, atom= 2765
Step= 16, Dmax= 3.9e-03 nm, Epot= 6.37647e+03 Fmax= 3.00818e+03, atom= 2765
Step= 17, Dmax= 4.6e-03 nm, Epot= 6.07244e+03 Fmax= 2.75324e+03, atom= 2765
Step= 19, Dmax= 2.8e-03 nm, Epot= 5.60144e+03 Fmax= 8.43337e+02, atom= 2765
Step= 20, Dmax= 3.3e-03 nm, Epot= 5.49136e+03 Fmax= 3.08327e+03, atom= 2765
Step= 21, Dmax= 4.0e-03 nm, Epot= 5.09456e+03 Fmax= 2.09083e+03, atom= 2765
Step= 23, Dmax= 2.4e-03 nm, Epot= 4.83141e+03 Fmax= 8.78173e+02, atom= 2765
Step= 25, Dmax= 1.4e-03 nm, Epot= 4.78063e+03 Fmax= 1.07042e+03, atom= 2765
Step= 26, Dmax= 1.7e-03 nm, Epot= 4.51750e+03 Fmax= 1.00741e+03, atom= 2765
Step= 27, Dmax= 2.1e-03 nm, Epot= 4.46707e+03 Fmax= 1.77657e+03, atom= 2765
Step= 28, Dmax= 2.5e-03 nm, Epot= 4.35788e+03 Fmax= 1.28547e+03, atom= 915
Step= 30, Dmax= 1.5e-03 nm, Epot= 4.20725e+03 Fmax= 6.94208e+02, atom= 915
Step= 31, Dmax= 1.8e-03 nm, Epot= 3.97280e+03 Fmax= 1.39504e+03, atom= 915
Step= 32, Dmax= 2.2e-03 nm, Epot= 3.97165e+03 Fmax= 1.44422e+03, atom= 915
Step= 33, Dmax= 2.6e-03 nm, Epot= 3.85030e+03 Fmax= 1.70904e+03, atom= 915
Step= 35, Dmax= 1.6e-03 nm, Epot= 3.71519e+03 Fmax= 3.29197e+02, atom= 472
Step= 36, Dmax= 1.9e-03 nm, Epot= 3.32876e+03 Fmax= 1.57250e+03, atom= 915
Step= 39, Dmax= 5.6e-04 nm, Epot= 3.23180e+03 Fmax= 8.45033e+02, atom= 2765
Step= 42, Dmax= 1.7e-04 nm, Epot= 3.20617e+03 Fmax= 6.21543e+02, atom= 915
Step= 50, Dmax= 1.6e-06 nm, Epot= 3.20591e+03 Fmax= 6.19379e+02, atom= 915
Step= 51, Dmax= 1.9e-06 nm, Epot= 3.20911e+03 Fmax= 6.16887e+02, atom= 2765
Stepsize too small, or no change in energy.
Converged to machine precision,
but not to the requested precision Fmax < 1
Double precision normally gives you higher accuracy.
writing lowest energy coordinates.
Steepest Descents converged to machine precision in 52 steps,
but did not reach the requested Fmax < 1.
Potential Energy = 3.2059077e+03
Maximum force = 6.1937860e+02 on atom 915
Norm of force = 1.7839124e+02
NOTE: 15 % of the run time was spent communicating energies,
you might want to use the -gcom option of mdrun
gcq#5: "We Can Dance Like Iggy Pop" (Red Hot Chili Peppers)
В ходе оптимизации геометрии максимальная сила изменилась на 3 порядка: от 4.37970e+05 на первом шаге до 6.16887e+02 на последнем (52) шаге.
Добавим в ячейку молекулы воды.
In [1]:
%%bash
genbox -cp b_em -p b -cs spc216 -o b_s
WARNING: Masses and atomic (Van der Waals) radii will be guessed
based on residue and atom names, since they could not be
definitively assigned from the information in your input
files. These guessed numbers might deviate from the mass
and radius of the atom type. Please check the output
files if necessary.
Neighborsearching with a cut-off of 0.45
Table routines are used for coulomb: FALSE
Table routines are used for vdw: FALSE
Cut-off's: NS: 0.45 Coulomb: 0.45 LJ: 0.45
System total charge: 0.000
Grid: 17 x 12 x 16 cells
:-) G R O M A C S (-:
Gromacs Runs On Most of All Computer Systems
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) genbox (-:
Option Filename Type Description
------------------------------------------------------------
-cp b_em.gro Input, Opt! Structure file: gro g96 pdb tpr etc.
-cs spc216.gro Input, Opt!, Lib. Structure file: gro g96 pdb tpr etc.
-ci insert.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-o b_s.gro Output Structure file: gro g96 pdb etc.
-p b.top In/Out, Opt! Topology file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-box vector 0 0 0 Box size
-nmol int 0 Number of extra molecules to insert
-try int 10 Try inserting -nmol times -try times
-seed int 1997 Random generator seed
-vdwd real 0.105 Default van der Waals distance
-shell real 0 Thickness of optional water layer around solute
-maxsol int 0 Maximum number of solvent molecules to add if
they fit in the box. If zero (default) this is
ignored
-[no]vel bool no Keep velocities from input solute and solvent
Reading solute configuration
bilayer in water
Containing 3200 atoms in 64 residues
Initialising van der waals distances...
Reading solvent configuration
"216H2O,WATJP01,SPC216,SPC-MODEL,300K,BOX(M)=1.86206NM,WFVG,MAR. 1984"
solvent configuration contains 648 atoms in 216 residues
Initialising van der waals distances...
Will generate new solvent configuration of 4x3x4 boxes
Generating configuration
Sorting configuration
Found 1 molecule type:
SOL ( 3 atoms): 10368 residues
Calculating Overlap...
box_margin = 0.315
Removed 12462 atoms that were outside the box
Successfully made neighbourlist
nri = 49430, nrj = 1740315
Checking Protein-Solvent overlap: tested 58585 pairs, removed 7842 atoms.
Checking Solvent-Solvent overlap: tested 124347 pairs, removed 2946 atoms.
Added 2618 molecules
Generated solvent containing 7854 atoms in 2618 residues
Writing generated configuration to b_s.gro
Back Off! I just backed up b_s.gro to ./#b_s.gro.1#
bilayer in water
Output configuration contains 11054 atoms in 2682 residues
Volume : 160.705 (nm^3)
Density : 920.108 (g/l)
Number of SOL molecules: 2618
Processing topology
Back Off! I just backed up b.top to ./#b.top.2#
gcq#318: "My Brothers are Protons (Protons!), My Sisters are Neurons (Neurons)" (Gogol Bordello)
Проведём "утряску" воды.
In [9]:
%%bash
grompp -f pr -c b_s -p b -o b_pr -maxwarn 1
mdrun -deffnm b_pr -v
:-) G R O M A C S (-:
GROup of MAchos and Cynical Suckers
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) grompp (-:
turning all bonds into constraints...
turning all bonds into constraints...
Analysing residue names:
There are: 64 Other residues
There are: 2618 Water residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Largest charge group radii for Van der Waals: 0.232, 0.232 nm
Largest charge group radii for Coulomb: 0.232, 0.232 nm
Calculating fourier grid dimensions for X Y Z
Using a fourier grid of 54x40x50, spacing 0.116 0.111 0.116
This run will generate roughly 2 Mb of data
Option Filename Type Description
------------------------------------------------------------
-f pr.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c b_s.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p b.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o b_pr.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]v bool no Be loud and noisy
-time real -1 Take frame at or first after this time.
-[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
-maxwarn int 1 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
-[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
-[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Ignoring obsolete mdp entry 'title'
Ignoring obsolete mdp entry 'cpp'
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.1#
NOTE 1 [file pr.mdp]:
nstcomm < nstcalcenergy defeats the purpose of nstcalcenergy, setting
nstcomm to nstcalcenergy
Generated 1369 of the 2211 non-bonded parameter combinations
Excluding 3 bonded neighbours molecule type 'DPPC'
Excluding 2 bonded neighbours molecule type 'SOL'
Velocities were taken from a Maxwell distribution at 300 K
Number of degrees of freedom in T-Coupling group DPPC is 6463.13
Number of degrees of freedom in T-Coupling group SOL is 15705.88
Estimate for the relative computational load of the PME mesh part: 0.42
There was 1 note
gcq#95: "Sort Of" (Urban Dance Squad)
:-) G R O M A C S (-:
GROup of MAchos and Cynical Suckers
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s b_pr.tpr Input Run input file: tpr tpb tpa
-o b_pr.trr Output Full precision trajectory: trr trj cpt
-x b_pr.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi b_pr.cpt Input, Opt. Checkpoint file
-cpo b_pr.cpt Output, Opt. Checkpoint file
-c b_pr.gro Output Structure file: gro g96 pdb etc.
-e b_pr.edr Output Energy file
-g b_pr.log Output Log file
-dhdl b_pr.xvg Output, Opt. xvgr/xmgr file
-field b_pr.xvg Output, Opt. xvgr/xmgr file
-table b_pr.xvg Input, Opt. xvgr/xmgr file
-tablep b_pr.xvg Input, Opt. xvgr/xmgr file
-tableb b_pr.xvg Input, Opt. xvgr/xmgr file
-rerun b_pr.xtc Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi b_pr.xvg Output, Opt. xvgr/xmgr file
-tpid b_pr.xvg Output, Opt. xvgr/xmgr file
-ei b_pr.edi Input, Opt. ED sampling input
-eo b_pr.edo Output, Opt. ED sampling output
-j b_pr.gct Input, Opt. General coupling stuff
-jo b_pr.gct Output, Opt. General coupling stuff
-ffout b_pr.xvg Output, Opt. xvgr/xmgr file
-devout b_pr.xvg Output, Opt. xvgr/xmgr file
-runav b_pr.xvg Output, Opt. xvgr/xmgr file
-px b_pr.xvg Output, Opt. xvgr/xmgr file
-pf b_pr.xvg Output, Opt. xvgr/xmgr file
-mtx b_pr.mtx Output, Opt. Hessian matrix
-dn b_pr.ndx Output, Opt. Index file
-multidir b_pr Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string b_pr Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 0 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Getting Loaded...
Reading file b_pr.tpr, VERSION 4.5.5 (single precision)
Starting 8 threads
Loaded with Money
Making 2D domain decomposition 4 x 1 x 2
starting mdrun 'bilayer in water'
1000 steps, 0.2 ps.
step 900, remaining runtime: 1 s imb F 20%
Writing final coordinates.
step 1000, remaining runtime: 0 s
Average load imbalance: 16.2 %
Part of the total run time spent waiting due to load imbalance: 1.9 %
Parallel run - timing based on wallclock.
NODE (s) Real (s) (%)
Time: 18.790 18.790 100.0
(Mnbf/s) (GFlops) (ns/day) (hour/ns)
Performance: 75.532 5.554 0.921 26.072
gcq#83: "All Beauty Must Die" (Nick Cave)
In [10]:
%%bash
editconf -f b_pr.gro -o b_pr.pdb
Read 11054 atoms Volume: 160.705 nm^3, corresponds to roughly 72300 electrons Velocities found
:-) G R O M A C S (-:
Gromacs Runs On Most of All Computer Systems
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) editconf (-:
Option Filename Type Description
------------------------------------------------------------
-f b_pr.gro Input Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-o b_pr.pdb Output, Opt! Structure file: gro g96 pdb etc.
-mead mead.pqr Output, Opt. Coordinate file for MEAD
-bf bfact.dat Input, Opt. Generic data file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-[no]ndef bool no Choose output from default index groups
-bt enum triclinic Box type for -box and -d: triclinic, cubic,
dodecahedron or octahedron
-box vector 0 0 0 Box vector lengths (a,b,c)
-angles vector 90 90 90 Angles between the box vectors (bc,ac,ab)
-d real 0 Distance between the solute and the box
-[no]c bool no Center molecule in box (implied by -box and -d)
-center vector 0 0 0 Coordinates of geometrical center
-aligncenter vector 0 0 0 Center of rotation for alignment
-align vector 0 0 0 Align to target vector
-translate vector 0 0 0 Translation
-rotate vector 0 0 0 Rotation around the X, Y and Z axes in degrees
-[no]princ bool no Orient molecule(s) along their principal axes
-scale vector 1 1 1 Scaling factor
-density real 1000 Density (g/L) of the output box achieved by
scaling
-[no]pbc bool no Remove the periodicity (make molecule whole again)
-resnr int -1 Renumber residues starting from resnr
-[no]grasp bool no Store the charge of the atom in the B-factor
field and the radius of the atom in the occupancy
field
-rvdw real 0.12 Default Van der Waals radius (in nm) if one can
not be found in the database or if no parameters
are present in the topology file
-[no]sig56 bool no Use rmin/2 (minimum in the Van der Waals
potential) rather than sigma/2
-[no]vdwread bool no Read the Van der Waals radii from the file
vdwradii.dat rather than computing the radii
based on the force field
-[no]atom bool no Force B-factor attachment per atom
-[no]legend bool no Make B-factor legend
-label string A Add chain label for all residues
-[no]conect bool no Add CONECT records to a .pdb file when written.
Can only be done when a topology is present
gcq#201: "Beat On the Brat With a Baseball Bat" (The Ramones)
In [11]:
%%bash
editconf -f b_s.gro -o b_s.pdb
Read 11054 atoms Volume: 160.705 nm^3, corresponds to roughly 72300 electrons No velocities found
:-) G R O M A C S (-:
S C A M O R G
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) editconf (-:
Option Filename Type Description
------------------------------------------------------------
-f b_s.gro Input Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-o b_s.pdb Output, Opt! Structure file: gro g96 pdb etc.
-mead mead.pqr Output, Opt. Coordinate file for MEAD
-bf bfact.dat Input, Opt. Generic data file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-[no]ndef bool no Choose output from default index groups
-bt enum triclinic Box type for -box and -d: triclinic, cubic,
dodecahedron or octahedron
-box vector 0 0 0 Box vector lengths (a,b,c)
-angles vector 90 90 90 Angles between the box vectors (bc,ac,ab)
-d real 0 Distance between the solute and the box
-[no]c bool no Center molecule in box (implied by -box and -d)
-center vector 0 0 0 Coordinates of geometrical center
-aligncenter vector 0 0 0 Center of rotation for alignment
-align vector 0 0 0 Align to target vector
-translate vector 0 0 0 Translation
-rotate vector 0 0 0 Rotation around the X, Y and Z axes in degrees
-[no]princ bool no Orient molecule(s) along their principal axes
-scale vector 1 1 1 Scaling factor
-density real 1000 Density (g/L) of the output box achieved by
scaling
-[no]pbc bool no Remove the periodicity (make molecule whole again)
-resnr int -1 Renumber residues starting from resnr
-[no]grasp bool no Store the charge of the atom in the B-factor
field and the radius of the atom in the occupancy
field
-rvdw real 0.12 Default Van der Waals radius (in nm) if one can
not be found in the database or if no parameters
are present in the topology file
-[no]sig56 bool no Use rmin/2 (minimum in the Van der Waals
potential) rather than sigma/2
-[no]vdwread bool no Read the Van der Waals radii from the file
vdwradii.dat rather than computing the radii
based on the force field
-[no]atom bool no Force B-factor attachment per atom
-[no]legend bool no Make B-factor legend
-label string A Add chain label for all residues
-[no]conect bool no Add CONECT records to a .pdb file when written.
Can only be done when a topology is present
gcq#179: "Have a Nice Day" (R. McDonald)
 |
 |
| Рис. 2а. До утряски воды. | Рис. 2b. После утряски воды. Липидные хвосты лежат теперь более свободно и хаотично. |
Далее запустили моделирование на суперкомпьютере.