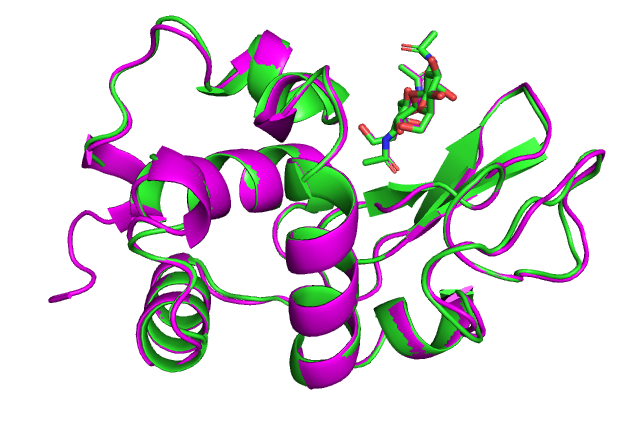
Гомологичное моделирование комплекса белка с лигандом
В этом задании будем моделировать комплекс лизоцима с лигандом.
In [1]:
import sys
sys.path.append('/usr/lib/modeller9v7/modlib/')
sys.path.append('/usr/lib/modeller9v7/lib/x86_64-intel8/python2.5/')
import modeller
import _modeller
import modeller.automodel
In [2]:
env=modeller.environ()
env.io.hetatm=True
MODELLER 9v7, 2009/06/12, r6923
PROTEIN STRUCTURE MODELLING BY SATISFACTION OF SPATIAL RESTRAINTS
Copyright(c) 1989-2009 Andrej Sali
All Rights Reserved
Written by A. Sali
with help from
B. Webb, M.S. Madhusudhan, M-Y. Shen, M.A. Marti-Renom,
N. Eswar, F. Alber, M. Topf, B. Oliva, A. Fiser,
R. Sanchez, B. Yerkovich, A. Badretdinov,
F. Melo, J.P. Overington, E. Feyfant
University of California, San Francisco, USA
Rockefeller University, New York, USA
Harvard University, Cambridge, USA
Imperial Cancer Research Fund, London, UK
Birkbeck College, University of London, London, UK
Kind, OS, HostName, Kernel, Processor: 4, Linux kodomo.fbb.msu.ru 3.8-2-amd64 x86_64
Date and time of compilation : 2009/06/12 12:23:44
MODELLER executable type : x86_64-intel8
Job starting time (YY/MM/DD HH:MM:SS): 2018/05/10 19:32:16
В качестве образца будем использовать известную структуру лизоцима из форели.
In [3]:
! wget http://www.pdb.org/pdb/files/1lmp.pdb
--2018-05-10 19:52:37-- http://www.pdb.org/pdb/files/1lmp.pdb
Resolving www.pdb.org... 132.249.213.195
Connecting to www.pdb.org|132.249.213.195|:80... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: https://www.rcsb.org/pdb/files/1lmp.pdb [following]
--2018-05-10 19:52:38-- https://www.rcsb.org/pdb/files/1lmp.pdb
Resolving www.rcsb.org... 132.249.213.190
Connecting to www.rcsb.org|132.249.213.190|:443... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: http://files.rcsb.org/view/1lmp.pdb [following]
--2018-05-10 19:52:39-- http://files.rcsb.org/view/1lmp.pdb
Resolving files.rcsb.org... 132.249.213.141
Connecting to files.rcsb.org|132.249.213.141|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: unspecified [text/plain]
Saving to: `1lmp.pdb'
[ <=> ] 130,410 126K/s in 1.0s
2018-05-10 19:52:40 (126 KB/s) - `1lmp.pdb' saved [130410]
А работать будем с лизоцимом из двустворчатого моллюска пэкхап, он же хамагури (Meretrix lusoria) (LYS_MERLU). У этого моллюска деликатесное нежное мясо, которое зимой едят сырым. Логично, у него же есть лизоцим, которым защищает от бактерий. Захотелось есть.
In [4]:
! wget http://www.uniprot.org/uniprot/P86383.fasta
--2018-05-10 20:24:52-- http://www.uniprot.org/uniprot/P86383.fasta Resolving www.uniprot.org... 193.62.193.81, 128.175.240.211 Connecting to www.uniprot.org|193.62.193.81|:80... connected. HTTP request sent, awaiting response... 200 OK Length: 194 [text/plain] Saving to: `P86383.fasta' 100%[======================================>] 194 --.-K/s in 0s 2018-05-10 20:24:52 (23.6 MB/s) - `P86383.fasta' saved [194/194]
In [22]:
alignm=modeller.alignment(env)
In [23]:
alignm.append(file='P86383.fasta', align_codes='all',alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
## есть смысл поправить идентификаторы
alignm[0].code = 'MERLU'
alignm[1].code = '1LMP'
In [24]:
alignm.salign()
alignm.write(file='all_in_one.ali', alignment_format='PIR')
SALIGN_____> adding the next group to the alignment; iteration 1
In [25]:
! cat all_in_one.ali
>P1;MERLU sequence:: : : : :::-1.00:-1.00 FAGGIVSQRCLSCICKMESGCRNVGCKMDMGSLSCGYFQIKEAYWIDCGRPGSSWKSCAASSYC--------ASL CVQNYMKRYAKWAGCPLRCEGFAREHNGGPRGCKKGSTIGYWNRLQKISGCHGVQ--* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGSTDYGIFQINSRYWCDDGRTPGAKNV CGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV...*
В последовательности лизоцима из моллюска не хватает жемчужины - лиганда. Добавим его.
In [37]:
with open("all_in_one.ali", 'r') as ali:
seq = ali.read().split("*")
seq[0] = seq[0][:-2] + "..."
seq[1] = seq[1][:-3] + "-..."
new = "*".join(seq)
with open("withlig.ali", 'w') as out:
out.write(new)
In [38]:
! cat withlig.ali
>P1;MERLU sequence:: : : : :::-1.00:-1.00 FAGGIVSQRCLSCICKMESGCRNVGCKMDMGSLSCGYFQIKEAYWIDCGRPGSSWKSCAASSYC--------ASL CVQNYMKRYAKWAGCPLRCEGFAREHNGGPRGCKKGSTIGYWNRLQKISGCHGVQ...* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGSTDYGIFQINSRYWCDDGRTPGAKNV CGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV-...*
In [39]:
## Выбираем объект для моделирования
s = alignm[0]
pdb = alignm[1]
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='withlig.ali', knowns= pdb.code, sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 2
a.make()
MERLU 1LMP
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 14 122
atom names : C +N
atom indices : 924 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 14 122
atom names : C CA +N O
atom indices : 924 918 0 925
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 1
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 11264 10431
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_484W> Dihedral still outside +-90: -90.1867
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 125
Number of all, selected real atoms : 969 969
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 10431 10431
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2153
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 695.1116
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 947 0 0 0.005 0.005 6.6637 1.000
2 Bond angle potential : 1266 0 2 1.982 1.982 97.908 1.000
3 Stereochemical cosine torsion poten: 591 0 31 49.004 49.004 211.46 1.000
4 Stereochemical improper torsion pot: 381 0 0 1.248 1.248 12.202 1.000
5 Soft-sphere overlap restraints : 2153 1 2 0.008 0.008 15.584 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2204 0 0 0.289 0.289 54.607 1.000
10 Distance restraints 2 (N-O) : 2337 0 0 0.347 0.347 80.981 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 121 0 0 4.036 4.036 23.247 1.000
14 Sidechain Chi_1 dihedral restraints: 97 0 1 86.038 86.038 29.467 1.000
15 Sidechain Chi_2 dihedral restraints: 65 0 0 87.077 87.077 26.006 1.000
16 Sidechain Chi_3 dihedral restraints: 33 0 0 83.699 83.699 23.143 1.000
17 Sidechain Chi_4 dihedral restraints: 17 0 0 85.250 85.250 9.9344 1.000
18 Disulfide distance restraints : 1 0 0 0.009 0.009 0.13882E-01 1.000
19 Disulfide angle restraints : 2 0 0 1.659 1.659 0.12148 1.000
20 Disulfide dihedral angle restraints: 1 0 0 36.120 36.120 1.1835 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 752 0 0 0.439 0.439 14.784 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 120 19 16 30.026 73.989 61.489 1.000
26 Distance restraints 4 (SDCH-SDCH) : 95 0 0 1.005 1.005 9.3167 1.000
27 Distance restraints 5 (X-Y) : 1401 0 0 0.038 0.038 16.997 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: MERLU.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 13777.8506
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3192 3G 4G C N 19 21 -85.42 -80.20 12.10 0.73 82.20 -131.92 7.47
1 4G 4G N CA 21 22 163.18 174.10 8.50
2 3203 14I 15C C N 98 100 -83.86 -63.00 69.30 7.34 -63.00 69.30 7.34
2 15C 15C N CA 100 101 24.99 -41.10 -41.10
3 3204 15C 16K C N 104 106 59.07 56.60 11.28 0.64 -62.90 139.84 24.43
3 16K 16K N CA 106 107 27.59 38.60 -40.80
4 3207 18E 19S C N 130 132 65.17 56.90 17.49 0.73 -64.10 140.88 18.80
4 19S 19S N CA 132 133 20.99 36.40 -35.00
5 3219 30M 31G C N 215 217 -63.67 -62.40 1.55 0.23 82.20 153.82 11.68
5 31G 31G N CA 217 218 -40.31 -41.20 8.50
6 3225 36G 37Y C N 249 251 65.33 55.90 9.84 0.94 -63.50 151.71 28.69
6 37Y 37Y N CA 251 252 36.71 39.50 -43.40
7 3226 37Y 38F C N 261 263 63.30 58.10 6.74 0.52 -63.20 146.01 26.29
7 38F 38F N CA 263 264 28.62 32.90 -44.30
8 3229 40I 41K C N 289 291 -106.73 -118.00 93.50 4.37 -62.90 97.49 11.20
8 41K 41K N CA 291 292 46.29 139.10 -40.80
9 3242 53S 54S C N 392 394 91.49 -136.60 136.62 4.58 -64.10 -151.88 17.72
9 54S 54S N CA 394 395 -173.21 151.20 -35.00
10 3245 56K 57S C N 421 423 55.46 56.90 17.41 0.99 -64.10 148.90 19.17
10 57S 57S N CA 423 424 53.75 36.40 -35.00
11 3254 65A 66S C N 478 480 157.33 56.90 103.86 10.66 -64.10 145.67 16.52
11 66S 66S N CA 480 481 9.92 36.40 -35.00
12 3256 67L 68C C N 492 494 -106.38 -117.90 41.59 1.44 -63.00 148.70 15.80
12 68C 68C N CA 494 495 101.13 141.10 -41.10
13 3264 75R 76Y C N 562 564 -98.66 -63.50 82.47 11.59 -63.50 82.47 11.59
13 76Y 76Y N CA 564 565 31.20 -43.40 -43.40
14 3269 80A 81G C N 607 609 19.51 78.70 89.51 1.80 -62.40 100.22 14.84
14 81G 81G N CA 609 610 -98.95 -166.10 -41.20
15 3282 93H 94N C N 708 710 -65.64 -119.90 54.26 2.25 -63.20 177.99 22.12
15 94N 94N N CA 710 711 136.87 137.00 -41.10
16 3283 94N 95G C N 716 718 123.33 82.20 60.35 1.82 -62.40 174.36 30.92
16 95G 95G N CA 718 719 -35.67 8.50 -41.20
17 3291 102K 103G C N 770 772 -62.91 -62.40 1.93 0.31 82.20 154.00 11.74
17 103G 103G N CA 772 773 -43.06 -41.20 8.50
18 3295 106I 107G C N 795 797 -74.40 -62.40 12.01 2.13 82.20 164.17 12.41
18 107G 107G N CA 797 798 -40.74 -41.20 8.50
19 3297 108Y 109W C N 811 813 52.93 58.80 10.72 0.30 -63.00 144.45 25.29
19 109W 109W N CA 813 814 41.97 33.00 -44.20
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 7 11 60 79 106 123 124 171 168 167
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 125
Number of all, selected real atoms : 969 969
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 10431 10431
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2185
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 859.9468
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 947 0 0 0.005 0.005 7.3707 1.000
2 Bond angle potential : 1266 2 5 3.045 3.045 140.37 1.000
3 Stereochemical cosine torsion poten: 591 0 32 50.116 50.116 220.55 1.000
4 Stereochemical improper torsion pot: 381 0 0 1.171 1.171 11.497 1.000
5 Soft-sphere overlap restraints : 2185 1 2 0.008 0.008 17.834 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2204 0 0 0.299 0.299 56.984 1.000
10 Distance restraints 2 (N-O) : 2337 0 4 0.439 0.439 133.31 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 121 0 1 4.150 4.150 24.576 1.000
14 Sidechain Chi_1 dihedral restraints: 97 0 2 89.468 89.468 38.168 1.000
15 Sidechain Chi_2 dihedral restraints: 65 0 0 92.713 92.713 29.983 1.000
16 Sidechain Chi_3 dihedral restraints: 33 0 0 77.922 77.922 21.701 1.000
17 Sidechain Chi_4 dihedral restraints: 17 0 0 88.084 88.084 11.568 1.000
18 Disulfide distance restraints : 1 0 0 0.012 0.012 0.23993E-01 1.000
19 Disulfide angle restraints : 2 0 0 1.376 1.376 0.83644E-01 1.000
20 Disulfide dihedral angle restraints: 1 0 0 33.469 33.469 1.0625 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 752 0 0 0.442 0.442 14.677 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 120 25 21 34.652 76.813 102.75 1.000
26 Distance restraints 4 (SDCH-SDCH) : 95 0 0 0.962 0.962 9.0394 1.000
27 Distance restraints 5 (X-Y) : 1401 0 0 0.040 0.040 18.397 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: MERLU.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 14940.5908
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3190 1F 2A C N 10 12 -66.99 -62.50 9.37 1.40 -134.00 -168.24 8.88
1 2A 2A N CA 12 13 -32.67 -40.90 147.00
2 3191 2A 3G C N 15 17 57.62 78.70 75.77 3.01 -80.20 147.66 7.73
2 3G 3G N CA 17 18 121.12 -166.10 174.10
3 3192 3G 4G C N 19 21 -81.75 -80.20 14.72 0.66 82.20 -137.13 7.31
3 4G 4G N CA 21 22 159.46 174.10 8.50
4 3194 5I 6V C N 31 33 -80.68 -125.40 51.80 2.76 -62.40 149.27 19.99
4 6V 6V N CA 33 34 169.45 143.30 -42.40
5 3195 6V 7S C N 38 40 75.94 56.90 78.56 3.60 -64.10 140.12 17.80
5 7S 7S N CA 40 41 -39.82 36.40 -35.00
6 3204 15C 16K C N 104 106 77.80 56.60 24.05 2.07 -62.90 156.28 27.30
6 16K 16K N CA 106 107 27.23 38.60 -40.80
7 3207 18E 19S C N 130 132 63.62 56.90 16.64 0.68 -64.10 139.53 18.61
7 19S 19S N CA 132 133 21.18 36.40 -35.00
8 3214 25G 26C C N 176 178 -56.05 -63.00 7.00 1.20 -117.90 -170.98 8.19
8 26C 26C N CA 178 179 -40.28 -41.10 141.10
9 3219 30M 31G C N 215 217 -63.22 -62.40 1.74 0.24 82.20 153.19 11.62
9 31G 31G N CA 217 218 -39.67 -41.20 8.50
10 3225 36G 37Y C N 249 251 65.27 55.90 11.08 0.84 -63.50 150.02 28.34
10 37Y 37Y N CA 251 252 33.57 39.50 -43.40
11 3226 37Y 38F C N 261 263 60.80 58.10 2.70 0.37 -63.20 146.00 26.32
11 38F 38F N CA 263 264 32.77 32.90 -44.30
12 3229 40I 41K C N 289 291 -101.67 -118.00 94.73 4.35 -62.90 94.87 10.92
12 41K 41K N CA 291 292 45.79 139.10 -40.80
13 3242 53S 54S C N 392 394 89.09 -136.60 139.00 4.67 -64.10 -153.82 17.45
13 54S 54S N CA 394 395 -173.00 151.20 -35.00
14 3245 56K 57S C N 421 423 55.52 56.90 15.02 0.84 -64.10 147.53 19.06
14 57S 57S N CA 423 424 51.35 36.40 -35.00
15 3254 65A 66S C N 478 480 157.15 56.90 105.98 10.33 -64.10 143.61 16.72
15 66S 66S N CA 480 481 2.04 36.40 -35.00
16 3256 67L 68C C N 492 494 -103.59 -117.90 45.74 1.56 -63.00 144.57 15.42
16 68C 68C N CA 494 495 97.65 141.10 -41.10
17 3264 75R 76Y C N 562 564 -97.96 -98.40 81.38 8.66 -63.50 96.77 13.80
17 76Y 76Y N CA 564 565 47.02 128.40 -43.40
18 3266 77A 78K C N 579 581 -65.32 -70.20 14.35 1.14 -62.90 165.33 21.60
18 78K 78K N CA 581 582 153.89 140.40 -40.80
19 3267 78K 79W C N 588 590 81.48 58.80 83.82 6.18 -71.30 154.86 10.36
19 79W 79W N CA 590 591 113.70 33.00 139.00
20 3269 80A 81G C N 607 609 24.81 78.70 86.30 1.60 -62.40 104.46 15.62
20 81G 81G N CA 609 610 -98.69 -166.10 -41.20
21 3291 102K 103G C N 770 772 -63.80 -62.40 2.13 0.39 82.20 154.76 11.79
21 103G 103G N CA 772 773 -42.81 -41.20 8.50
22 3294 105T 106I C N 787 789 -128.12 -120.60 60.52 3.75 -63.40 130.96 19.93
22 106I 106I N CA 789 790 70.25 130.30 -43.60
23 3297 108Y 109W C N 811 813 64.25 58.80 9.87 0.28 -63.00 144.74 25.61
23 109W 109W N CA 813 814 24.77 33.00 -44.20
24 3300 111R 112L C N 844 846 -118.87 -108.50 91.69 5.06 -63.50 99.44 12.25
24 112L 112L N CA 846 847 41.39 132.50 -41.20
25 3305 116S 117G C N 884 886 56.94 78.70 24.15 0.87 -80.20 140.45 10.07
25 117G 117G N CA 886 887 -155.62 -166.10 174.10
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 11 11 60 83 128 112 149 140 166 172
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
MERLU.B99990001.pdb 695.11157
MERLU.B99990002.pdb 859.94684
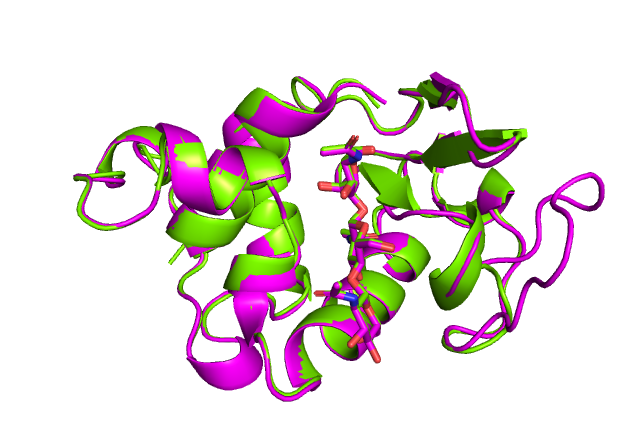
Теперь поместим лиганд в другое место.
In [40]:
! rm MERLU.rsr
In [44]:
class mymodel(modeller.automodel.automodel):
def special_restraints(self, aln):
rsr = self.restraints
at = self.atoms
for x,y in [('O6:123', 'CG:83')]:
rsr.add(modeller.forms.gaussian(group=modeller.physical.xy_distance,
feature=modeller.features.distance(at[x],at[y]),
mean=3.0, stdev=0.1))
from modeller import *
from modeller.automodel import *
a = mymodel(env, alnfile='withlig.ali', knowns= pdb.code, sequence = s.code)
a.make()
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 14 122
atom names : C +N
atom indices : 924 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 14 122
atom names : C CA +N O
atom indices : 924 918 0 925
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 1
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 11265 10432
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 125
Number of all, selected real atoms : 969 969
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 10432 10432
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2321
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 2104.5730
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 947 3 5 0.016 0.016 61.470 1.000
2 Bond angle potential : 1266 4 16 2.811 2.811 190.95 1.000
3 Stereochemical cosine torsion poten: 591 0 33 49.281 49.281 221.46 1.000
4 Stereochemical improper torsion pot: 381 0 1 1.498 1.498 18.718 1.000
5 Soft-sphere overlap restraints : 2321 1 2 0.009 0.009 19.998 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2204 9 37 0.633 0.633 390.74 1.000
10 Distance restraints 2 (N-O) : 2337 3 29 0.605 0.605 332.53 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 121 2 5 6.436 6.436 59.106 1.000
14 Sidechain Chi_1 dihedral restraints: 97 0 2 88.757 88.757 42.508 1.000
15 Sidechain Chi_2 dihedral restraints: 65 0 1 86.191 86.191 27.168 1.000
16 Sidechain Chi_3 dihedral restraints: 33 0 0 77.983 77.983 22.387 1.000
17 Sidechain Chi_4 dihedral restraints: 17 0 0 95.604 95.604 12.628 1.000
18 Disulfide distance restraints : 1 0 0 0.014 0.014 0.35678E-01 1.000
19 Disulfide angle restraints : 2 0 0 0.911 0.911 0.36676E-01 1.000
20 Disulfide dihedral angle restraints: 1 0 0 5.003 5.003 0.30699E-01 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 752 0 0 0.374 0.374 13.444 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 120 29 24 36.903 79.132 137.64 1.000
26 Distance restraints 4 (SDCH-SDCH) : 95 0 0 0.908 0.908 7.6630 1.000
27 Distance restraints 5 (X-Y) : 1402 3 13 0.148 0.148 546.06 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: MERLU.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 17945.7656
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 1 : Bond length potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 637 83P 83P CA CB 620 622 1.71 1.53 0.18 5.04 1.53 0.18 5.04
2 639 83P 83P CG CD 623 621 1.80 1.54 0.26 7.09 1.54 0.26 7.09
3 640 83P 83P N CD 619 621 1.64 1.46 0.18 5.92 1.46 0.18 5.92
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1797 83P 83P N CA 619 620 128.92 108.20 20.73 4.71 108.20 20.73 4.71
-------------------------------------------------------------------------------------------------
Feature 9 : Distance restraints 1 (CA-CA)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4834 40I 83P CA CA 284 620 11.82 8.98 2.84 4.81 8.98 2.84 4.81
2 5520 75R 83P CA CA 554 620 7.58 5.09 2.49 6.76 5.09 2.49 6.76
3 5559 80A 83P CA CA 605 620 7.50 5.22 2.28 5.80 5.22 2.28 5.80
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 7523 83P 77A N O 619 580 12.69 7.30 5.38 6.57 7.30 5.38 6.57
2 7526 83P 80A N O 619 608 5.33 3.14 2.18 4.79 3.14 2.18 4.79
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3192 3G 4G C N 19 21 -82.91 -80.20 10.27 0.54 82.20 -133.06 7.34
1 4G 4G N CA 21 22 164.19 174.10 8.50
2 3202 13C 14I C N 90 92 -95.72 -120.60 84.16 3.90 -63.40 98.93 15.28
2 14I 14I N CA 92 93 49.90 130.30 -43.60
3 3203 14I 15C C N 98 100 -147.98 -63.00 103.85 12.98 -63.00 103.85 12.98
3 15C 15C N CA 100 101 18.59 -41.10 -41.10
4 3204 15C 16K C N 104 106 54.47 56.60 5.17 0.53 -62.90 139.12 24.25
4 16K 16K N CA 106 107 33.89 38.60 -40.80
5 3207 18E 19S C N 130 132 59.79 56.90 18.65 0.96 -64.10 134.75 17.99
5 19S 19S N CA 132 133 17.98 36.40 -35.00
6 3209 20G 21C C N 140 142 57.12 -69.10 144.31 13.42 -117.90 -171.26 7.62
6 21C 21C N CA 142 143 -148.25 141.80 141.10
7 3210 21C 22R C N 146 148 -75.83 -63.00 16.16 2.01 57.30 150.07 18.64
7 22R 22R N CA 148 149 -31.26 -41.10 38.00
8 3219 30M 31G C N 215 217 -64.78 -62.40 4.43 0.60 82.20 154.00 11.64
8 31G 31G N CA 217 218 -37.47 -41.20 8.50
9 3225 36G 37Y C N 249 251 66.97 55.90 13.08 0.99 -63.50 150.96 28.50
9 37Y 37Y N CA 251 252 32.54 39.50 -43.40
10 3226 37Y 38F C N 261 263 61.85 58.10 4.52 0.39 -63.20 145.65 26.24
10 38F 38F N CA 263 264 30.38 32.90 -44.30
11 3229 40I 41K C N 289 291 -96.99 -62.90 84.26 9.70 -62.90 84.26 9.70
11 41K 41K N CA 291 292 36.25 -40.80 -40.80
12 3242 53S 54S C N 392 394 94.81 -136.60 132.03 4.52 -64.10 -145.64 18.10
12 54S 54S N CA 394 395 -178.86 151.20 -35.00
13 3244 55W 56K C N 412 414 -106.53 -62.90 79.52 9.20 -62.90 79.52 9.20
13 56K 56K N CA 414 415 25.68 -40.80 -40.80
14 3245 56K 57S C N 421 423 45.00 56.90 25.64 1.07 -64.10 144.09 18.18
14 57S 57S N CA 423 424 59.11 36.40 -35.00
15 3254 65A 66S C N 478 480 80.09 56.90 25.92 2.26 -64.10 156.12 20.85
15 66S 66S N CA 480 481 24.83 36.40 -35.00
16 3256 67L 68C C N 492 494 -88.23 -117.90 40.68 1.12 -63.00 156.42 17.52
16 68C 68C N CA 494 495 113.27 141.10 -41.10
17 3264 75R 76Y C N 562 564 -99.10 -63.50 78.69 11.00 -63.50 78.69 11.00
17 76Y 76Y N CA 564 565 26.78 -43.40 -43.40
18 3265 76Y 77A C N 574 576 -66.30 -62.50 20.83 3.61 -134.00 166.05 10.25
18 77A 77A N CA 576 577 -61.38 -40.90 147.00
19 3266 77A 78K C N 579 581 137.60 56.60 121.66 7.31 -62.90 159.91 26.67
19 78K 78K N CA 581 582 -52.18 38.60 -40.80
20 3270 81G 82C C N 611 613 -63.34 -63.00 94.56 11.56 -63.00 94.56 11.56
20 82C 82C N CA 613 614 -135.65 -41.10 -41.10
21 3271 82C 83P C N 617 619 -55.41 -64.50 81.87 5.65 -58.70 96.39 8.23
21 83P 83P N CA 619 620 65.84 147.20 -30.50
22 3272 83P 84L C N 624 626 -93.78 -63.50 35.55 6.55 -63.50 35.55 6.55
22 84L 84L N CA 626 627 -59.83 -41.20 -41.20
23 3282 93H 94N C N 708 710 -63.35 -119.90 56.57 2.38 -63.20 179.59 22.44
23 94N 94N N CA 710 711 138.49 137.00 -41.10
24 3283 94N 95G C N 716 718 126.29 82.20 64.35 1.95 -62.40 171.33 30.49
24 95G 95G N CA 718 719 -38.37 8.50 -41.20
25 3291 102K 103G C N 770 772 -62.22 -62.40 1.96 0.28 82.20 153.38 11.70
25 103G 103G N CA 772 773 -43.16 -41.20 8.50
26 3295 106I 107G C N 795 797 -75.05 -62.40 12.76 2.34 82.20 165.44 12.54
26 107G 107G N CA 797 798 -42.92 -41.20 8.50
27 3297 108Y 109W C N 811 813 67.98 58.80 17.42 0.48 -63.00 145.08 25.70
27 109W 109W N CA 813 814 18.20 33.00 -44.20
28 3300 111R 112L C N 844 846 -113.62 -108.50 90.66 4.95 -63.50 97.12 11.92
28 112L 112L N CA 846 847 41.99 132.50 -41.20
29 3306 117G 118C C N 888 890 69.07 57.40 33.91 1.09 -117.90 -139.34 9.59
29 118C 118C N CA 890 891 4.17 36.00 141.10
-------------------------------------------------------------------------------------------------
Feature 27 : Distance restraints 5 (X-Y)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 10432 123. 83P O6 CG 939 623 6.72 3.00 3.72 37.23 3.00 3.72 37.23
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 1 11 18 59 81 133 130 138 157 181 153
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
MERLU.B99990001.pdb 2104.57300
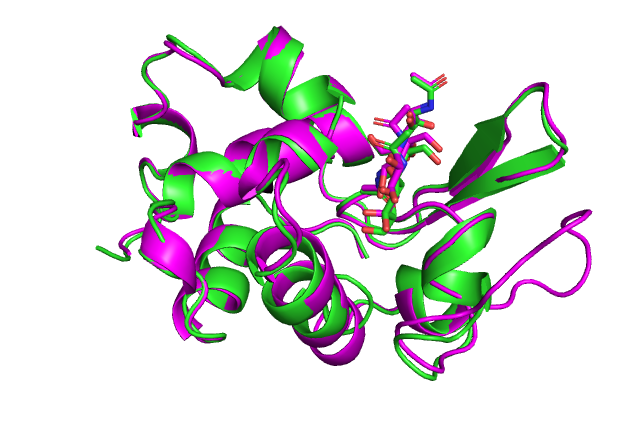
А теперь заменим в лизоциме все аминокислоты на аланин и снова построим комплекс с лигандом.
In [51]:
with open("P86383.fasta", 'r') as fasta:
strs = fasta.read().split("1\n")
prot = strs[1].replace("\n", "")
ala = "A"*len(prot)
with open("alala.fasta", 'w') as alala:
alala.write(strs[0] + "1\n" + ala + "\n")
In [52]:
alignm=modeller.alignment(env)
alignm.append(file='alala.fasta', align_codes='all',alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
alignm[0].code = 'ALA'
alignm[1].code = '1LMP'
alignm.salign()
alignm.write(file='aliala.ali', alignment_format='PIR')
SALIGN_____> adding the next group to the alignment; iteration 1
In [53]:
! cat aliala.ali
>P1;ALA sequence:: : : : :::-1.00:-1.00 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA----------* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGSTDYGIFQINSRYWCDDGRTPGAKNV CGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV...*
In [54]:
with open("aliala.ali", 'r') as ali:
seq = ali.read().split("*")
seq[0] = seq[0][:-3] + "..."
new = "*".join(seq)
with open("aliala.ali", 'w') as out:
out.write(new)
In [55]:
! cat aliala.ali
>P1;ALA sequence:: : : : :::-1.00:-1.00 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA-------...* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGSTDYGIFQINSRYWCDDGRTPGAKNV CGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV...*
In [56]:
## Выбираем объект для моделирования
s = alignm[0]
pdb = alignm[1]
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='aliala.ali', knowns= pdb.code, sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 2
a.make()
ALA 1LMP
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 1 122
atom names : C +N
atom indices : 609 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 1 122
atom names : C CA +N O
atom indices : 609 607 0 610
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 9565 8962
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 125
Number of all, selected real atoms : 654 654
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 8962 8962
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 1145
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 555.0652
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 610 0 0 0.006 0.006 6.5517 1.000
2 Bond angle potential : 853 0 5 2.231 2.231 86.531 1.000
3 Stereochemical cosine torsion poten: 248 0 30 76.553 76.553 235.37 1.000
4 Stereochemical improper torsion pot: 244 0 0 0.922 0.922 5.4153 1.000
5 Soft-sphere overlap restraints : 1145 1 2 0.010 0.010 14.430 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2232 0 0 0.203 0.203 36.672 1.000
10 Distance restraints 2 (N-O) : 2385 0 2 0.305 0.305 87.188 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 121 0 3 4.553 4.553 29.584 1.000
14 Sidechain Chi_1 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
15 Sidechain Chi_2 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
16 Sidechain Chi_3 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
17 Sidechain Chi_4 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
18 Disulfide distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
19 Disulfide angle restraints : 0 0 0 0.000 0.000 0.0000 1.000
20 Disulfide dihedral angle restraints: 0 0 0 0.000 0.000 0.0000 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 693 0 0 0.226 0.226 4.4737 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 120 17 15 27.952 61.264 33.999 1.000
26 Distance restraints 4 (SDCH-SDCH) : 55 0 0 0.327 0.327 0.71275 1.000
27 Distance restraints 5 (X-Y) : 1401 0 0 0.034 0.034 14.134 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: ALA.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 10055.5547
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1970 15A 16A C N 74 76 78.29 55.40 26.03 1.99 -62.50 155.80 31.66
1 16A 16A N CA 76 77 25.81 38.20 -40.90
2 1973 18A 19A C N 89 91 65.57 55.40 23.97 0.87 -62.50 140.33 28.50
2 19A 19A N CA 91 92 16.49 38.20 -40.90
3 1975 20A 21A C N 99 101 53.66 55.40 4.56 0.42 -62.50 138.21 28.08
3 21A 21A N CA 101 102 33.99 38.20 -40.90
4 1976 21A 22A C N 104 106 63.13 55.40 11.50 0.56 -62.50 144.09 29.30
4 22A 22A N CA 106 107 29.68 38.20 -40.90
5 1991 36A 37A C N 179 181 67.70 55.40 15.29 0.99 -62.50 147.83 30.07
5 37A 37A N CA 181 182 29.12 38.20 -40.90
6 1992 37A 38A C N 184 186 62.52 55.40 12.09 0.51 -62.50 142.95 29.07
6 38A 38A N CA 186 187 28.43 38.20 -40.90
7 2003 48A 49A C N 239 241 85.71 55.40 41.10 2.31 -62.50 156.85 31.69
7 49A 49A N CA 241 242 10.44 38.20 -40.90
8 2008 53A 54A C N 264 266 88.83 -134.00 143.09 3.46 -62.50 -159.61 31.49
8 54A 54A N CA 266 267 -172.25 147.00 -40.90
9 2011 56A 57A C N 279 281 61.43 55.40 12.47 1.23 -62.50 153.17 31.04
9 57A 57A N CA 281 282 49.11 38.20 -40.90
10 2021 66A 67A C N 329 331 81.19 55.40 61.25 2.21 -62.50 145.60 28.84
10 67A 67A N CA 331 332 -17.35 38.20 -40.90
11 2024 69A 70A C N 344 346 -63.20 -62.50 82.99 13.64 -68.20 90.95 7.51
11 70A 70A N CA 346 347 -123.89 -40.90 145.30
12 2028 73A 74A C N 364 366 53.68 55.40 22.57 1.22 -62.50 154.34 31.06
12 74A 74A N CA 366 367 60.70 38.20 -40.90
13 2031 76A 77A C N 379 381 64.91 55.40 10.41 1.24 -62.50 152.23 30.92
13 77A 77A N CA 381 382 42.42 38.20 -40.90
14 2058 103A 104A C N 514 516 81.91 -134.00 158.87 3.61 -62.50 178.67 28.71
14 104A 104A N CA 516 517 -146.11 147.00 -40.90
15 2071 116A 117A C N 579 581 56.03 55.40 6.66 0.35 -62.50 138.92 28.24
15 117A 117A N CA 581 582 31.57 38.20 -40.90
16 2074 119A 120A C N 594 596 -101.87 -68.20 53.99 5.28 -62.50 149.29 22.88
16 120A 120A N CA 596 597 103.10 145.30 -40.90
17 2075 120A 121A C N 599 601 -122.49 -134.00 14.45 0.37 -62.50 -171.07 28.69
17 121A 121A N CA 601 602 138.25 147.00 -40.90
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 4 4 25 46 59 64 66 93 91 93
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 125
Number of all, selected real atoms : 654 654
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 8962 8962
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 1177
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 858.7646
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 610 0 0 0.007 0.007 8.4452 1.000
2 Bond angle potential : 853 1 10 2.678 2.678 125.78 1.000
3 Stereochemical cosine torsion poten: 248 0 29 75.452 75.452 230.40 1.000
4 Stereochemical improper torsion pot: 244 0 0 1.148 1.148 8.5706 1.000
5 Soft-sphere overlap restraints : 1177 1 2 0.011 0.011 16.387 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2232 0 2 0.286 0.286 95.506 1.000
10 Distance restraints 2 (N-O) : 2385 0 15 0.427 0.427 186.39 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 121 1 6 6.302 6.302 56.677 1.000
14 Sidechain Chi_1 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
15 Sidechain Chi_2 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
16 Sidechain Chi_3 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
17 Sidechain Chi_4 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
18 Disulfide distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
19 Disulfide angle restraints : 0 0 0 0.000 0.000 0.0000 1.000
20 Disulfide dihedral angle restraints: 0 0 0 0.000 0.000 0.0000 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 693 0 0 0.338 0.338 10.955 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 120 22 19 34.901 67.129 99.026 1.000
26 Distance restraints 4 (SDCH-SDCH) : 55 0 0 0.721 0.721 3.3601 1.000
27 Distance restraints 5 (X-Y) : 1401 0 0 0.038 0.038 17.273 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: ALA.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 11366.2793
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1113 72A 73A C N 359 361 141.13 120.00 21.13 4.80 120.00 21.13 4.80
-------------------------------------------------------------------------------------------------
Feature 13 : Mainchain Omega dihedral restraints
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2148 73A 73A CA C 362 364 -143.08 -180.00 36.92 7.38 -180.00 36.92 7.38
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1957 2A 3A C N 9 11 -66.88 -62.50 4.42 0.87 -68.20 173.18 14.01
1 3A 3A N CA 11 12 -41.52 -40.90 145.30
2 1958 3A 4A C N 14 16 68.89 -68.20 138.31 11.08 -68.20 138.31 11.08
2 4A 4A N CA 16 17 126.96 145.30 145.30
3 1970 15A 16A C N 74 76 77.48 55.40 24.05 2.00 -62.50 156.32 31.78
3 16A 16A N CA 76 77 28.67 38.20 -40.90
4 1972 17A 18A C N 84 86 -55.10 -62.50 7.53 1.38 -134.00 -171.97 11.60
4 18A 18A N CA 86 87 -42.32 -40.90 147.00
5 1974 19A 20A C N 94 96 48.40 -68.20 123.51 11.65 -68.20 123.51 11.65
5 20A 20A N CA 96 97 -173.96 145.30 145.30
6 1975 20A 21A C N 99 101 -80.84 -68.20 53.72 4.76 -62.50 135.24 21.34
6 21A 21A N CA 101 102 93.09 145.30 -40.90
7 1976 21A 22A C N 104 106 65.61 55.40 29.75 1.12 -62.50 137.95 27.96
7 22A 22A N CA 106 107 10.25 38.20 -40.90
8 1991 36A 37A C N 179 181 65.80 55.40 10.72 1.02 -62.50 149.37 30.37
8 37A 37A N CA 181 182 35.60 38.20 -40.90
9 1992 37A 38A C N 184 186 61.21 55.40 14.03 0.51 -62.50 140.37 28.55
9 38A 38A N CA 186 187 25.43 38.20 -40.90
10 2003 48A 49A C N 239 241 81.86 55.40 34.36 2.07 -62.50 155.27 31.46
10 49A 49A N CA 241 242 16.28 38.20 -40.90
11 2008 53A 54A C N 264 266 94.08 -134.00 137.42 3.33 -62.50 -154.17 32.41
11 54A 54A N CA 266 267 -174.50 147.00 -40.90
12 2011 56A 57A C N 279 281 61.33 55.40 12.23 1.21 -62.50 152.95 31.00
12 57A 57A N CA 281 282 48.89 38.20 -40.90
13 2021 66A 67A C N 329 331 78.40 55.40 56.86 2.06 -62.50 143.48 28.54
13 67A 67A N CA 331 332 -13.80 38.20 -40.90
14 2025 70A 71A C N 349 351 63.12 55.40 8.02 0.94 -62.50 149.61 30.39
14 71A 71A N CA 351 352 40.36 38.20 -40.90
15 2027 72A 73A C N 359 361 -80.60 -62.50 44.93 6.75 -62.50 44.93 6.75
15 73A 73A N CA 361 362 0.22 -40.90 -40.90
16 2028 73A 74A C N 364 366 -22.96 -62.50 40.99 7.35 -62.50 40.99 7.35
16 74A 74A N CA 366 367 -51.71 -40.90 -40.90
17 2029 74A 75A C N 369 371 76.85 -62.50 146.75 25.92 -62.50 146.75 25.92
17 75A 75A N CA 371 372 -86.91 -40.90 -40.90
18 2031 76A 77A C N 379 381 169.79 -134.00 86.37 2.85 -62.50 166.31 33.54
18 77A 77A N CA 381 382 -147.43 147.00 -40.90
19 2032 77A 78A C N 384 386 -72.73 -62.50 32.55 5.88 -134.00 153.92 9.50
19 78A 78A N CA 386 387 -71.80 -40.90 147.00
20 2058 103A 104A C N 514 516 80.06 -134.00 159.42 3.63 -62.50 178.82 28.59
20 104A 104A N CA 516 517 -148.86 147.00 -40.90
21 2071 116A 117A C N 579 581 51.51 55.40 8.00 0.30 -62.50 142.87 28.92
21 117A 117A N CA 581 582 45.20 38.20 -40.90
22 2075 120A 121A C N 599 601 -57.32 -68.20 13.07 0.86 -62.50 179.02 29.59
22 121A 121A N CA 601 602 138.05 145.30 -40.90
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 8 3 31 48 62 57 70 91 103 82
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
ALA.B99990001.pdb 555.06525
ALA.B99990002.pdb 858.76459