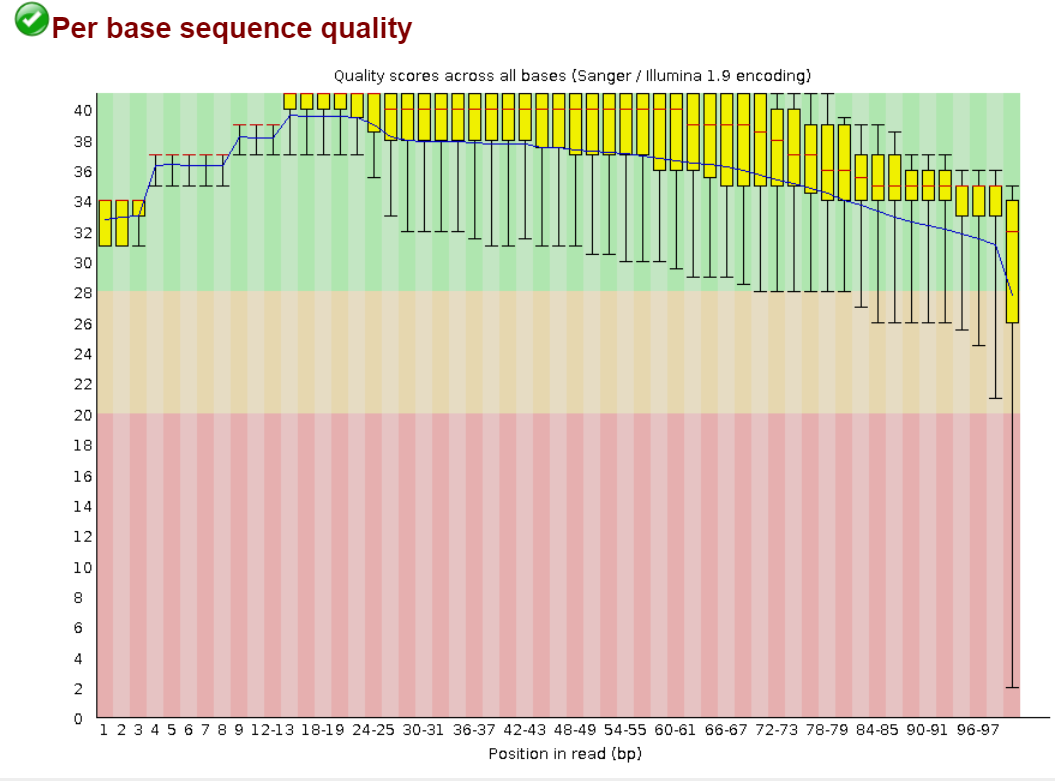
Рис.1. Контроль качества чтений до чистки |
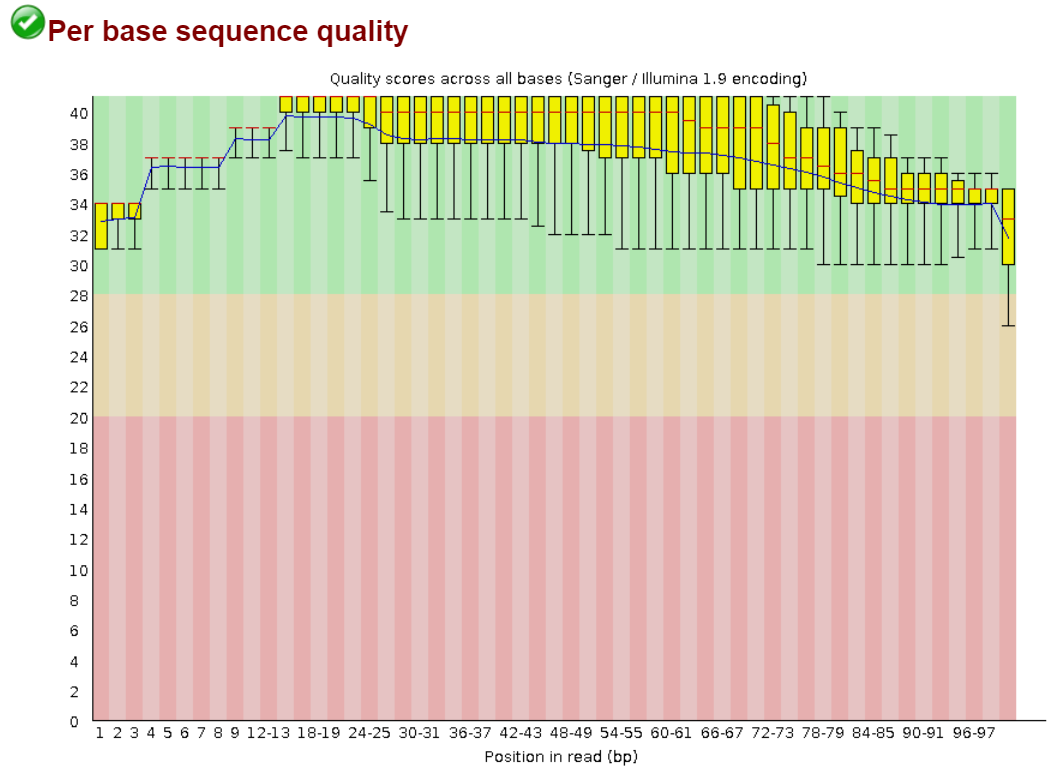
Рис.2. Контроль качества чтений после чистки |
В данном практикуме мы работали с чтениями экзома, картирующимися на участок хромосомы человека.
Файл chr3.fastq с одноконцевыми чтениями в формате fastq был взят мной из директории /P/y14/term3/block4/SNP/reads.
Подготовка чтений осуществлялась в несколько этапов:
1. Анализ качества прочтений
2. Очистка чтений с последующим анализом и сравнением параметров прочтений до и после очистки
Анализ прочтений был осуществлен с помощью прогрммы fastQC, установленной на сервере kodomo. Результатом работы программы является zip архив и html файл, содержащий отчет о качестве прочтений.
Программа была запущена с помощью команды:
fastqc chr3.fastq
Анализ прочтений был осуществлен с помощью прогрммы fastQC, установленной на сервере kodomo. Результатом работы программы является zip архив и html файл, содержащий отчет о качестве прочтений.
Программа была запущена с помощью команды:
fastqc chr3.fastq
Очистка чтений проводилась с использованием программы Trimmomatic. Были удалены чтения с длиной менее 50 нуклеотидов и с концов каждого чтения были отрезаны нуклеотиды с качеством ниже 20.
Ниже приведена команда:
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr3.fastq chr3_out.fastq TRAILING:20 MINLEN:50
Сравним параметры чтений до и после чистки. До чистки было 20932 чтений, после чистки стало 20570 чтений.
На рис.1 изображен контроль качества чтений до чистки, а на рис.2 - после чистки. Синяя линия - среднее качество чтений, центральные красные линии - медианы, желтые прямоугольники - интерквартальные размахи (разница между верхн. и нижн. квартилями; диапазон значений качества, при котором качество 25% чтений на данной позиции выше нижней границы, а 75% - не выше верхней).
Поле графика разделено на 3 полосы - зеленую, желтую и красную; попадание в данные полосы вышеперечисленных элементов графика позволяет сделать вывод о качестве чтений. После чистки все риды располагаются в зеленой области, следовательно, с помощью чистки мы получили надежные прочтения. Также после чистки стало меньше ридов, так как были удалены риды длинной меньше 50.
|
Рис.1. Контроль качества чтений до чистки |
Рис.2. Контроль качества чтений после чистки |
Чтения были откартированы с помощью программы hisat2. Программа была импортирована с помощью команды:
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
| Команда | Функция | Выдача |
| hisat2-build chr3.fasta chr3 | Индексирование референсной последовательности | Индексированный файл chr3.fasta |
| hisat2 -x chr3 -U chr3_out.fastq --no-spliced-alignment --no-softclip > align_pr11.sam | Выравнивание чтений после чистки с референсной последовательностью | Файл, который содержит выравнивание формата SAM align_pr11.sam |
Табл.1. Команды, использованные при картировании последовательности.
Программа картирования была запущена с параметрами -x (путь к индексу) -U (путь к чтениям) --no-spliced-alignment (запрет на картирование с разрывами) --no-softclip (картирование без подрезания чтений).
Также необходимо было проанализировать полученное выравнивание. Для этого я использовала программу Samtools. Она работает с файлами SAM формата.
| Команда | Функция | Выдача |
| samtools view align_pr11.sam -bo align_pr11.bam | Программа переводит файл в формат bam | align_pr11.bam |
| samtools sort align_pr11.bam -T sort.txt -o sort.bam | Сортировка выравнивания чтений и референса по координате в референсе | sort.bam |
| samtools index sort.bam | Индексирование отсортированного выравнивания | sort.bam |
| samtools idxstats sort.bam > sumread.txt | Запись числа откартировавшихся чтений | sumread.txt |
Табл.2. Команды, использованные для анализа последовательностей в формате SAM.
Оказалось, что на хромосому откартировалось 20510 ридов. 79 ридов не было откартировано вообще.
Запись числа откартировавшихся ридов| Команда | Функция | Выдача |
| samtools mpileup -uf chr3.fasta sort.bam > snp.bcf | Создание файла с полиморфизмами | snp.bcf |
| bcftools call -cv snp.bcf -o snp.vcf | Определение различий | snp.vcf |
Табл.3. Команды, использованные при поиске SNP и инделей.
В результате были найдены 230 SNP. Из них были выбраны 3 штуки и описаны в таблице.
| Координата | Тип полиморфизма | В референсе | В чтениях | Покрытие в этом месте | Качество покрытия |
| chr 3:41291081 | замена | G | A | 25 | 221.999 |
| chr 3:41831203 | замена | C | T | 30 | 206.009 |
| chr 3:41309334 | замена | C | A | 3 | 4.12848 |
Табл.4. Найденные SNP и индели.
Далее необходимо было аннотировать полученные SNP. Аннотирование осуществлялось с помощью программы ANNOVAR.
Подготовка входных файлов включала удаление инделей из snp.vcf и использование скрипта convert2annovar.pl
Использованная команда: perl /nfs/srv/databases/annovar/convert2annovar.pl.old -format vcf4 /nfs/srv/databases/ngs/ciara_mak/snp.vcf > /nfs/srv/databases/ngs/ciara_mak/snp.avinput
Далее провелась аннотация:
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1. rs.snp.hg19_snp138_dropped - содержит полиморфизмы, имеющие rs в dbsnp, то есть аннотированные в этой базе данных.
2. rs.snp.hg19_snp138_filtered- содержит отфильтрованные полиморфизмы, то есть, не имеющие rs в dbsnp.
3. rs.dbsnp.log - содержит отчет о работе команды.
Аннотация по базе dbsnp показала, что 177 полиморфизмов имеют rs, а 41 не имеют.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.1000genomes -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1. rs.1000g.hg19_ALL.sites.2014_10_dropped - содержит полиморфизмы, имеющие rs в 1000 genomes, и их частоты.
2. rs.1000g.hg19_ALL.sites.2014_10_filtered -содержит полиморфизмы, не имеющие rs в 1000 genomes.
3. rs.1000.log - содержит отчет о работе команды.
Аннотация по базе 1000 genomes показала, что 174 полиморфизмов имеют rs, а 44 не имеют. Дополнительно мы смогли узнать частоты аннотированных полиморфизмов.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1. rs.gwas.hg19_gwasCatalog_dropped - содержит SNP, имеющие известное клиническое значение.
Файл оказался пустым, то есть аннотация не дала никаких результатов.
2. rs.gwas.hg19_gwasCatalog_filtered- содержит полиморфизмы, не имеющие rs в GWAS.
3. rs.gwas.log - содержит отчет о работе команды.
Аннотация по базе GWAS показала, что 218 полиморфизмов не имеют rs.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.clinvar -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1. rs.clinvar.hg19_clinvar_20150629_dropped - содержит SNP, имеющие известное клиническое значение.
Файл оказался пустым, то есть аннотация не дала никаких результатов.
2. rs.clinvar.hg19_clinvar_20150629_filtered - содержит полиморфизмы, не имеющие rs в Clinvar.
3. rs.clinvar.log - содержит отчет о работе команды.
Аннотация по базе Clinvar показала, что 218 полиморфизмов не имеют rs.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1. rs.refgene.variant_function - содержит описание всех полиморфизмов.
2. rs.refgene.exonic_variant_function - содержит описание полиморфизмов внутри экзонов.
3. rs.refgene.log - содержит отчет о работе команды.
В данном случае SNP подразделяются на несколько категорий, которые отображены в первой колонке файла rs.refgene.variant_function. Они указывают, где находится данный полиморфизм - внутри экзона, интрона, гена некодирующей РНК и т.д.
Возможные категории:
exonic - полиморфизм внутри экзона (частично или полностью)
splicing - полиморфизм в пределах 2 bp от границы сплайсинга (число bp можно изменить)
ncRNA - полиморфизм полностью или частично входит в транскрипт, не имеющий аннотации как кодирующий
UTR5 - полиморфизм полностью или частично входит в 5-нетранслируемую область
UTR3 - полиморфизм полностью или частично входит в 3-нетранслируемую область
intronic - полиморфизм полностью или частично внутри интрона
downstream - полиморфизм в пределах 1-kb downstream от сайта окончания транскрипции (параметр может быть изменен)
upstream - полиморфизм в пределах 1-kb upstream от сайта начала транскрипции (параметр может быть изменен)
intergenic - полиморфизм на пересечении генов
В моём случае количество SNP: 198 intronic, 13 exonic, 2 UTR5, 4 UTR3, 1 intergenic
| Ген | Число SNP |
|---|---|
| ULK4 | 161 |
| CCK | 1 |
| LYZL4 | 1 |
| GNL3 | 3 |
| CADM2 | 2 |
| FNDC3B | 51 |
© Макиевская Кьяра, 2018