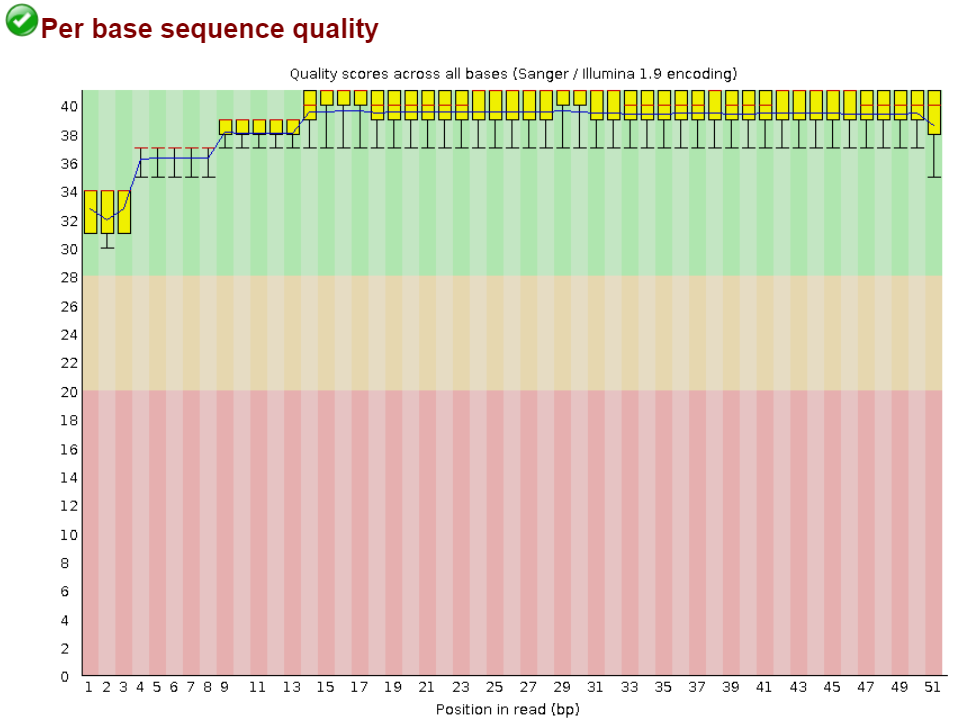
Рис.1. Контроль качества чтений до чистки |
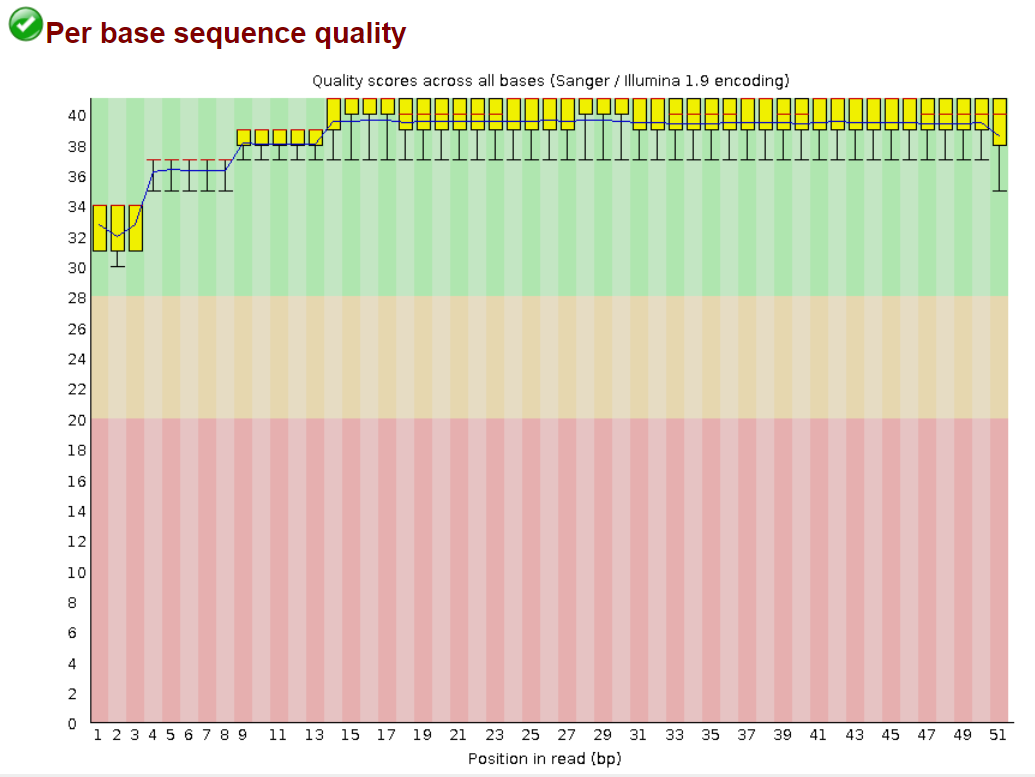
Рис.2. Контроль качества чтений после чистки |
Была создана директория /nfs/srv/databases/ngs/ciara_mak/pr12, в которой выполнялись все задания практикума. Туда были скопированы файл с последовательностью 3 хромосомы (chr3.fasta) и файлы с одноконцевыми чтениями в формате fastq (chr3.1.fastaq и chr3.2.fastaq), однако дальше задания выполнялись с использованием chr3.1.fastaq.
Далее с помощью программы FastQC было установлено качество прочтений. Результатом работы программы был html файл и zip архив.
Команда запуска программы: fastqc chr3.1.fastq
Далее при помощи программы Trimmomatiс были удалены чтения с длиной менее 50 нуклеотидов и с концов каждого чтения были отрезаны нуклеотиды с качеством ниже 20.
Команда запуска программы: java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr3.1.fastq chr3.1_out.fastq TRAILING:20 MINLEN:50
До чистки было 21211 чтений, после чистки стало 21152 чтений.
На рис.1 изображен контроль качества чтений до чистки. Синяя линия на графике - среднее качество чтений, центральные красные линии - медианы, желтые прямоугольники - интерквартальные размахи (разница между верхней и нижней квартилями, диапазон значений качества, при котором качество 25% чтений на данной позиции выше нижней границы, а 75% - не выше верхней). Поле графика разделено на 3 полосы зеленого, желтого и красного цветов, попадание в которые вышеперечисленных элементов графика позволяет судить о качестве чтений.
На рис.2 изображен контроль качества чтений после чистки. До чистки чтения были хорошие, поэтому после нее мы не видим особых изменений. С помощью чистки мы получили самые надежные прочтения длинной меньше 50.
|
Рис.1. Контроль качества чтений до чистки |
Рис.2. Контроль качества чтений после чистки |
Была создана директория /nfs/srv/databases/ngs/ciara_mak/pr12, в которой выполнялись все задания практикума. Туда были скопированы файлы с одноконцевыми чтениями в формате fastq (chr3.1.fastaq и chr3.2.fastaq), однако дальше задания выполнялись с использованием chr3.1.fastaq.
Далее с помощью программы FastQC было установлено качество прочтений. Результатом работы программы был html файл и zip архив.
Команда запуска программы: fastqc chr3.1.fastq
Далее при помощи программы Trimmomatiс были удалены чтения с длиной менее 50 нуклеотидов и с концов каждого чтения были отрезаны нуклеотиды с качеством ниже 20.
Команда запуска программы: java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr3.1.fastq chr3.1_out.fastq TRAILING:20 MINLEN:50
До чистки было 21211 чтений, после чистки стало 21152 чтений.
На рис.1 изображен контроль качества чтений до чистки. Синяя линия на графике - среднее качество чтений, центральные красные линии - медианы, желтые прямоугольники - интерквартальные размахи (разница между верхней и нижней квартилями, диапазон значений качества, при котором качество 25% чтений на данной позиции выше нижней границы, а 75% - не выше верхней). Поле графика разделено на 3 полосы зеленого, желтого и красного цветов, попадание в которые вышеперечисленных элементов графика позволяет судить о качестве чтений.
На рис.2 изображен контроль качества чтений после чистки. До чистки чтения были хорошие, поэтому после нее мы не видим особых изменений. С помощью чистки мы получили самые надежные прочтения длинной меньше 50.
Чтения были откартированы с помощью программы hisat2 аналогично тому, как это проводилось в практикуме 11.
Программа была импортирована с помощью команды: export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
| Команда | Функция | Выдача |
| hisat2-build chr3.fasta chr3 | Индексирование референсной последовательности | Индексированный файл chr3.fasta |
| hisat2 -x chr3 -U chr3.1_out.fastq --no-softclip > chr3.sam | Выравнивание чтений после чистки с референсной последовательностью | Файл, который содержит выравнивание формата SAM chr3.sam |
Табл.1. Команды, использованные при картировании последовательности.
Мы не использовали параметр, запрещающий без разрывов картировать, так как в данном случае у нас данные транскриптомного анализа, то есть мы работаем с РНК. По сравнению с ДНК с ней могли произойти модификации, связанные с перегруппировкой (сплайсинг).
Далее необходимо было проанализировать полученное выравнивание. Для этого я использовала программу Samtools. Она работает с файлами в формате SAM.
| Команда | Функция | Выдача |
| samtools view chr3.sam -bo chr3.bam | Программа переводит файл в формат bam | chr3.bam |
| samtools sort chr3.bam -T nexact.txt -o chr3_sort.bam | Сортировка выравнивания чтений и референса по координате в референсе | chr3_sort.bam |
| samtools index chr3_sort.bam | Индексирование отсортированного выравнивания | chr3_sort.bam |
| samtools idxstats chr3_sort.bam > result.txt | Запись числа откартировавшихся чтений | result.txt |
Табл.2. Команды, использованные для анализа последовательностей в формате SAM.
Выяснилось, что на хромосому откартировалось 21019 рида. 154 рида не были откартированы.
Далее осуществлялся подсчет чтений.
Команда запуска программы: htseq-count -f bam chr3_sort.bam -m union -i gene_id -s no /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseq.txt
Опции программы:
-f формат bam или sam
-s цепь прямая (yes) или обратная (no)
-i атрибут gff
-m режим обработки перекрытия ридов
"Ненулевые" строки были посчитаны с помощью grep'а
Выдача:
ENSG00000072274.8 - 20495
ENSG00000252174.1 - 4
__no_feature - 478
__not_aligned - 154
__alignment_not_unique - 42
Не все чтения, а именно 478 ридов не легли в границы генов, еще 42 рида по режиму union пересекаются сразу с несколькими генами.
Доставшийся ген TFR1 (ENSG00000072274.8) - ген рецептора трансферрина человека, необходимый для эндоцитоза ионов железа из трансферрина в цитоплазму клетки. Другие 4 пересечения (ENSG00000252174.1) ассоциированы с неэкспрессирующимся псевдогеном.
© Макиевская Кьяра, 2018