Выравнивания
Jalview
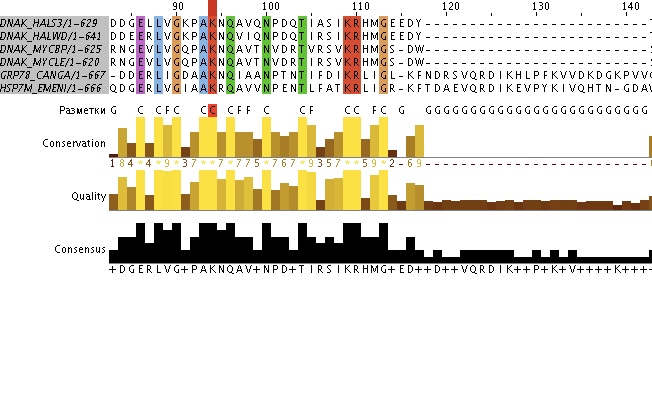
Рисунок справа содержит фрагмент выравнивания белков семейства HSP70, выбранных мной.
Для рисунка была выбрана раскраска Clustalx с параметром идентичности 100%. В поле "Разметка" содержится информация о сходности аминокислот в колонках. C - абсолютно консервативные, F - функционально консервативные, G - в этом месте расположен геп, что означает делецию, или инсерцию.
Немного о консервативности.
Для изучения консервативности нами были взяты те же 6 белков из нескольких крупных таксонов живых организмов. Archaea: DNAK_HALS3, DNAK_HALWD; Bacteria: DNAK_MYCBP, DNAK_MYCLE; Eucaryota: HSP7M_EMENI, GRP78_CANGA. Выравнивание называется top1.fasta.
Рассчёты проводились с помощью infoalign, программы пакета EMBOSS, которая способна подсчитывать количество совпадений входных последовательностей и консенсусной последовательностии по какому-то параметру. Мы использовали такие параметры: наличие гэпов, наличие абсолютно консервативных позиций (АКП), наличие функционально консервативных позиций (ФКП), наличие позиций, консервативных на 70% (70%КП). Стоит отметить, что длина консенсусной последовательности составляет 723 а.о.
| Название | Длина | Кол. гэпов | Прц. гэпов | Кол. АКП | Прц. АКП | Кол. ФКП | Прц. ФКП | Кол. 70%КП | Прц. 70%КП | |
| DNAK_HALS3_1 | 629 | 94 | 13,00% | 196 | 27,11% | 333 | 46,06% | 279 | 38,59% | |
| DNAK_HALWD_1 | 641 | 82 | 11,34% | 196 | 27,11% | 333 | 46,06% | 277 | 38,31% | |
| DNAK_MYCBP_1 | 625 | 98 | 13,55% | 196 | 27,11% | 333 | 46,06% | 271 | 37,48% | |
| DNAK_MYCLE_1 | 620 | 103 | 14,25% | 196 | 27,11% | 333 | 46,06% | 271 | 37,48% | |
| GRP78_CANGA_1 | 667 | 56 | 7,75% | 196 | 27,11% | 333 | 46,06% | 261 | 36,10% | |
| HSP7M_EMENI_1 | 666 | 57 | 7,88% | 196 | 27,11% | 333 | 46,06% | 254 | 35,13% |
Как можно видеть на рисунке и в таблице, которая содержит информацию, полученную из infoalign, это семейство белков крайне не консервативно. Лишь 27% участков во всех шести белках имеют одинаковый аминокислотный состав. Но этот параметр позволяет лишь ужасаться, а делать выводы по нему рано. Важнее рассмотреть ФКП, которые отражают наличие позиций, в которых хоть мутация и произошла, но она не сильно повлияла на функции. К примеру, во всех позициях F, обозначенных на рисунке, представлены только гидрофобные аминокислоты, что и позволяет их объединить по функциональности. Таких позиций в выравнивании около 40-50%. Но этот параметр тоже довольно интересен для группировки организмов. Также можно отметить, что по этому параметру все организмы довольно чётко разбиваются на группы, что говорит возможном изменении белка уже у общих предков этих организмов (для каждой из трёх групп один предок). И последний интересный факт можно выявить из данных в таблице (на рисунке тоже): эукариотические белки довольно резко отличаются и от бактериальных, и от архибактериальных. Они имеют большую длину (примерно 7% гэпов у них и 13% гэпов у бактерий с археями), а так же на рисунке заметно большее аминокислотное сходство белков первых четырёх организмов.
В общем такие различия понятны. Белки данного семейства шаперонов выполняют хоть и сходные функции, однако, они должны уметь взаимодействовать со слишком различными белковыми системами, которые могут иметь большое различие у эукариот и прокариот.
Странная эволюция
Настоящей эволюции это соответствует мало, так что просто представлю таблицу с мутациями.
| Номер | Тип мутации | Расположение | Поколение |
| 1 | Ins A | 4 | p7-p8 |
| 2 | Del V | 6 | p3-p4 |
| 3 | Del N | 12 | p1-p2 |
| 4 | Del V | 17 | p1-p2 |
| 5 | Ins M | 17 | p6-p7 |
| 6 | Rep G-A | 20 | p4-p5 |
| 7 | Ins E | 25 | p6-p7 |
| 8 | Ins R | 27 | p3-p4 |
| 9 | Rep I-F | 28 | p5-p6 |
| 10 | Rep F-T | 28 | p7-p8 |
А теперь перейдём к рассмотрению того, зачем же мы исправляли некоторые места самостоятельно. Алгоритм выравнивания Tcoffee, котрый я исползовал, стреится уменьшить количество гэпов в последовательности, из-за чего периодически случаются ошибки, вес которых меньше, чем вес гэпа. Но мы знаем алгоритмы, которыми сеяли мутации в последовательностях. Вследствие чего можем исправлять мутации до их количества 7мт/поколение.

К примеру, изначально в четвёртой позиции на местах гэпов стояли G, что увеличивало количество мутаций p7-p8 на одну. Вставка гэпа в том месте уменьшила их количество, вследствие чего это поколение снова попадало в 7мт/поколение. Или E, расположенные в 34 позиции в p3 и p4, до редакции находились в позиции 31, что можно обосновать сходством их свойств, вследствие чего алгоритм не придал этому значения(данные выравнивания называются fastagodU.fasta - без редакции, fastagodR.fasta - с редакцией).
В эксперименте, описанном выше, всё было логично и понятно. Но это не совсем относилось к 3 эксперименту. Точечные мутации в ДНК вносят много проблем. Во-первых в последовательностях часто образовывались стоп-кодоны, из за чего позиции выпадали из состава транскриптов. Другая проблема - инделы приводят к свигу рамок считывания, что коренным обазом влияет на последовательность белка. Однако, единственным спасавшим нас обстоятельством являлось то, что кодоны, полученные при замене третьего нуклеотида, транслруют (практически всегда) одинаковые аминокислоты. Вследствие этого после постройки последовательностей белков НЕ ПРОИЗВОДИЛОСЬ ручное выравнивание, так как по оно не несло бы никакой смысловой нагрузки. Необходимо было произвести выравнивание по последовательностям ДНК, что не являлось нашей задачей (данное выравнивание называется fastagod2U.fasta). Эволюция была построена с помощью topscript2.sh и topscript3.sh, выходным файлом из которых является fastagod.fasta. Также они оставляют после себя файлы последовательностей белков и аминокислот каждого из этапов. В первом случае на вход подавать ничего не нужно. Во втором случае принимается файл sequence.fasta с последовательностью ДНК.
О презентации...
Всё будет перечислено в хаотичном беспорядке, но по пунктам. Сначала немного пройдёмся по самой презентации, раз уж такое задание:
- Слайд 6: рибозимы также умеют читать нуклеотидные последовательности (на этом строится одна из теорий о происхождении жизни;
- Слайд 12: однояйцевые близнецы, несмотря на то, что являются производными одной гаметы, имеют всё же отличающиеся последовательности ДНК, так как наблюдаются мутации при репликациях ДНК.
- Мутации происходят не всегда случайно. В некоторых местах имеются системы, увеличивающие их количество (имунная система).
- Наследуются мутации во всех клетках зародышевого пути, так как линия зародышевого пути бессмертна и непрерывна.
- Не всегда последовательности белка под стабилизирующим отбором. Те же белки рецепторов Т-лимфоцитов не стабилизируются
- И не всегда можно по структуре сказать о том, что белки гомологичны. То есть не все белки с одинаковой последовательность
Скачать все файлы практикума
Выравнивание 1 (полное)
Не хотелось загромождать страницу, поэтому, оно здесь.
© Попов Алексей, 2016 г.
