Гомологичное моделирование комплекса белка с лигандом
В этом задании было проведено моделирование белка LYS_BPAPS.
1) Выравнивание
С помощью ClustalW было построено выравнивание последовательностей моего белка и белка 1LMP:
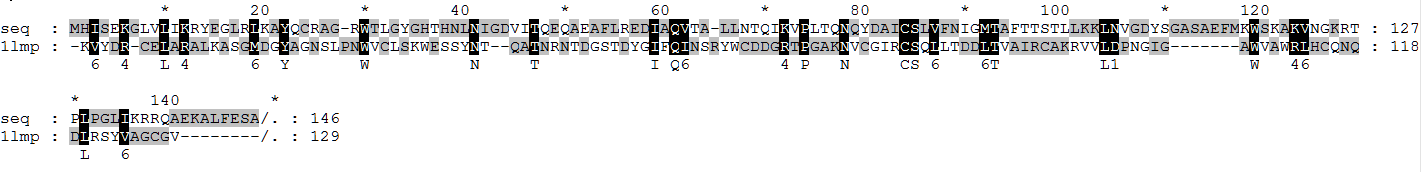
В качестве формата выходного файла был указан PIR.
2) Модификация файла выравнивания
Переименовала последовательность в файле выравнивания:
| Было |
Стало |
| >P1;uniprot|P37712|LYSC_CAMDR |
>P1;seq |
| >P1;1LMP|PDBID|CHAIN|SEQUENCE |
>P1;1lmp |
После имени последовательности моделируемого белка надо добавить строчку:
sequence:ХХХХХ::::::: 0.00: 0.00
эта строчка описывает входные параметры последовательности для modeller. После имени последовательности белка-образца добавить:
structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00
эта строчка описывает, какой файл содержит структуру белка с этой последовательностью,
номера первой и последней аминокислот в структуре, идентификатор цепи и т.д.
В конце каждой последовательности надо добавить символы:
/.
Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки).
Модифицированное выравнивание формата PIR
3) Модификация файла со структурой
Удалила всю воду из структуры, в итоге получился файл 1lmp_now.ent
4) Создала скрипт lys_bpaps.py, в котором указано:
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4);
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема);
атомы белка, образующие водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов (длина данного белка 226 а.о., а белка-модели - 146,
поэтому номера а.о. лиганда изменились);
параметры взаимного расположения атомов пары описаны в строчке 9-10. 3 точки могут однозначно расположить сложную структуру в пространстве,
поэтому мы выбираем водородные связи как источник данных точек;
что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами, строчка 12;
что имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании);
что число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15
что пора строить модель, строчка 16
Запустила исполнение скрипта.
Получила 5 моделей, лучшей из которых, на мой взгляд, оказалась такая:
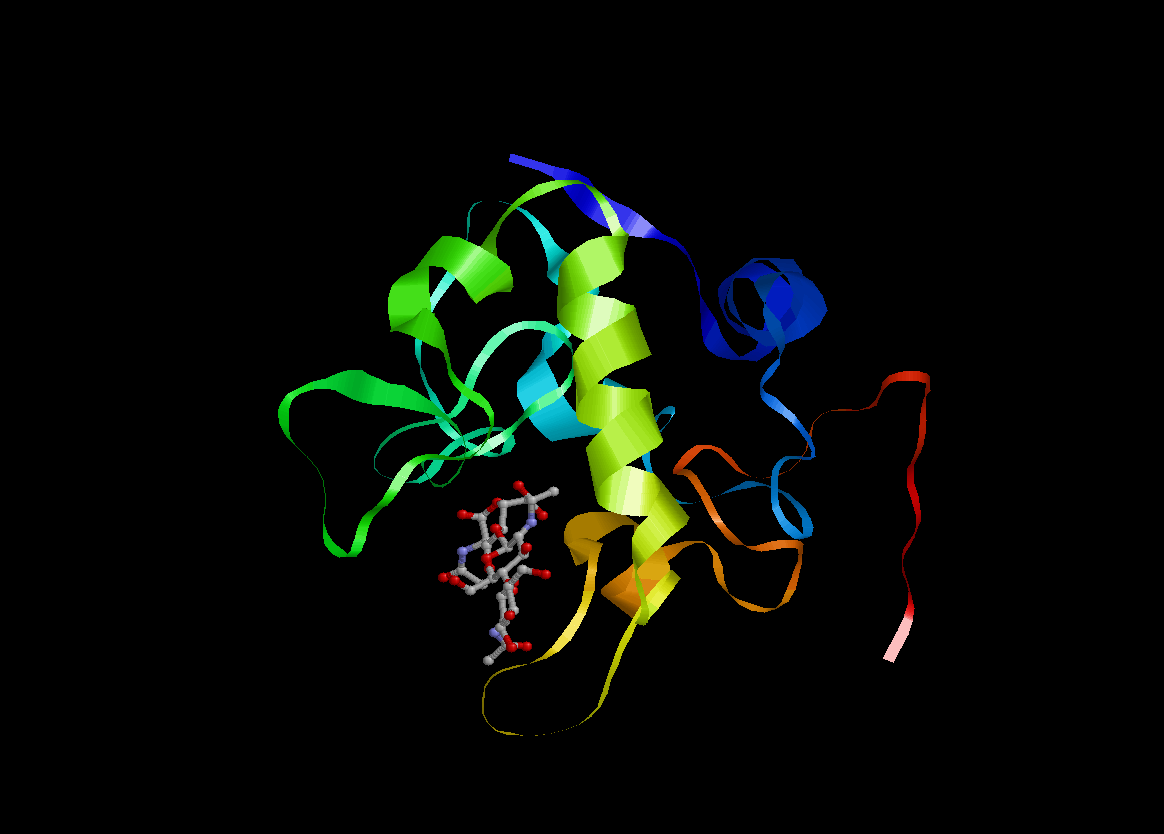
5) Анализ результатов
Выбор наилучшей модели был основан на 2-х критериях: Omega и Ramachandran Z-score
a) Omega
1 - Omega average and std. deviation= 180.886 8.177 Significant deviations from expected 5.5!!!
2 - Omega average and std. deviation= 180.259 7.752
Significant deviations from expected 5.5!!!
3 - Omega average and std. deviation= 180.850 7.696
Significant deviations from expected 5.5!!!
4 - Omega average and std. deviation= 180.628 8.918
Significant deviations from expected 5.5!!!
5 - Omega average and std. deviation= 180.212 7.735
Significant deviations from expected 5.5!!!
1LMP - Omega average and std. deviation= 179.870 2.718 Significant deviations from expected 5.5!!
б) Ramachandran Z-score
1 - (-3.381)
2 - (-2.837)
3 - (-2.641)
4 - (-3.553)
5 - (-3.852)
1LMP - (-0.965)
По обоим параметрам лучшей оказалась модель №3.
© Свистунова Даша