
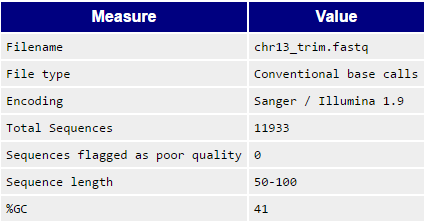
Мне была дана 13 хромосома человека chr13.fasta и, соответственно, файл с ридами для той же хромосомы chr13.fastq (без адаптеров).
Сперва я выполнила анализ качества чтений с помощью программы FastQC. Я воспользовалась версией, установленной на сервере kodomo. В командной строке я написала: fastqc chr13.fastq
Программа выдала архив файлов chr13_fastqc.zip и отчет о работе программы chr13_fastqc.html.
Очистка чтений была выполнена с помощью программы Trimmomatic (версии, установленной на kodomo).
Использовавшаяся команда:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr13.fastq chr13_out.fastq TRAILING:20
MINLEN:50
Где TRAILING - удаляет с конца чтения нуклеотиды с качеством ниже указанного, а MINLEN - удаляет чтения с длиной меньше указанного числа.
В результате был получен очищенный файл chr13_trim.fastq, который был проанализирован с помощью программы FastQC.
Использовавшаяся команда:
fastqc chr13_out.fastq
На выходе получился файл с отчётом chr13_trim_fastqc.html.
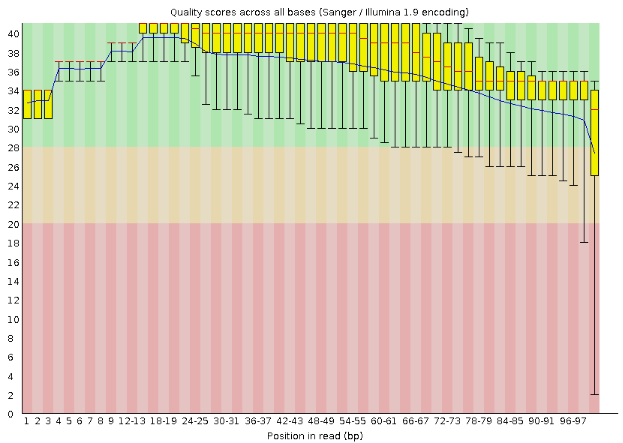
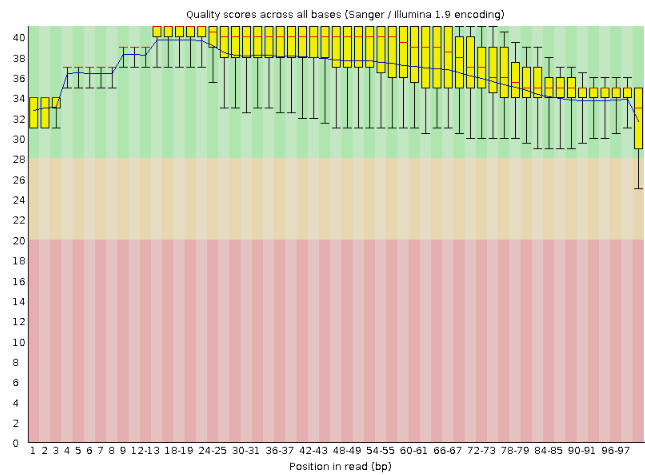
Характеристики чтения до(слева)/после(справа) очистки
| |
| |
Per base sequence quality для последовательности приемлемо и до чистки, и после (программа не выдает предупреждение). Также видно, что после чистки больше нет "усов" (края статистически значимой выборки) выходящих из зеленой области, некоторые интерквартильные промежутки (желтые прямоугольники) уменьшились, математическое ожидание по качеству (синяя линия) повысилось. То есть качество чтений увеличилось. Таким образом, были вырезаны только низкокачественные чтения и остались чтения пригодные для работы.
Сперва было необходимо проиндексировать референсную последовательность. Для этого я воспользовалась программой BWA (Burrows-Wheeler Aligner).
Так как она установлена на сервере kodomo, то я запустила её, введя в командной строке:
bwa index chr13.fasta
Затем было построено выравнивание очищенных прочтений и референса в формате .sam по алгоритму mem. Для этого была использована команда:
bwa mem chr13.fasta chr13_trim.fastq > chr13.sam
На выходе я получила файл chr13.sam.
Теперь мне нужно было перевести его в бинарный формат .bam. Для этого я воспользовалась следующей командой пакета samtools:
samtools view chr13.sam -b -o chr13.bam
Опция -b меняет формат файла на .bam, а опция -o указывает на название выходного файла. В результате я получила файл chr13.bam.
Затем я отсортировала получившийся файл по координате начала чтения в референсе.
Для этого я использовала команду:
samtools sort chr13.bam -T t.txt -o chr13_sort.bam
Опция -T позволяет записывать временные файлы в temp.txt, а не в stdout, а опция -o указывает на название выходного файла. В результате я получила отсортированный файл chr13_sort.bam. Затем было необходимо его проиндексировать, для чего я воспользовалась командой:
samtools index chr13_sort.bam
Результат: индексный файл chr13_sort.bam.bai. Для того, чтобы узнать сколько чтений откартировалось на геном, я воспользовалась командой:
samtools idxstats chr13_sort.bam > result.txt
В результате получен файл results.txt, в котором указаны название последовательности, длина последовательности (115169878) и число картированных чтений (11932). Получается, что все чтения были картированы.
| Команда | Функция | Результат |
|---|---|---|
| samtools mpileup -uf chr13.fasta chr13_sort.bam -o snp.bcf | Создание файла с полиморфизмами в формате bcf | snp.bcf |
| bcftools call -cv snp.bcf -o snp.vcf | Создание файла со списком отличий между референсом и чтениями в формате vcf | snp.vcf |
Всего в файле snp.vcf указано 137 полиморфизмов, из них 6 вставок, 7 делеций и 122 замен. Покрытие и качество полиморфизмов встречается как хорошее (например качество 225.009 и покрытие 225), так и не очень (например качество 4.12848 и покрытие 3). Среднее значение качества - 77,76, покрытия - 17,42. В целом показатели достаточно нормальные.
| Примеры полиморфизмов | |||||
|---|---|---|---|---|---|
| Координата | Тип | В референсе | В прочтении | Качество прочтения на участке | Глубина покрытия на участке |
| 25516889 | Замена | G | A | 147,008 | 22 |
| 25527657 | Делеция | GAAAA | GAAA | 54,4663 | 45 |
| 25511130 | Вставка | T | TCA | 165,469 | 9 |
Далее необходимо было аннотировать полученные SNP с помощью программы ANNOVAR. Для работы с программой, файл необходимо можифицировать. В первую очередь из файла snp.vcf были удалены все индели, полученный файл - snp_mod.vcf. Затем был использован скрипт convert2annovar.pl для получения файла snp.avinput:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_mod.vcf -outfile snp.avinput
Далее аннтоции были получены с помощью скрипта annotate_variation.pl
Refgene - база данных для метода аннотации полиморфизмов, основанного на изменении строении генов и межгенных участков при инделях или заменах. Программа выдает 3 файла: в первом (variant_function) суммарная информация о всех найденных SNP, во втором (exonic_variant_function) содержится информация о SNP в экзонах, а в третем (log) информация о процессе работы программы.
В результате работы скрипта полученны файлы: chr13.refgene.variant_function; chr13.refgene.exonic_variant_function ; chr13.refgene.log. Файл chr13.refgene.variant_function содержит описание всех полиморфизмов, в первой колонке содержатся категории snp, которые указывают на то, где находится полиморфизм (exonic-внутри экзона, intronic-внутри интрона и т. д.). А также в файле указано гетерозиготный (52) или гомозиготный (72) полиморфизм.
| exonic | intronic | UTR3 | ncRNA_intronic | ncRNA_exonic | upstream | downstream | Всего |
| 7 | 78 | 6 | 34 | 3 | 1 | 1 | 35 |
Категории:
В категории, для которых указывается ген, попали 124 полиморфизма. Все они расположены на одном из двух генов - PHF11 или COL4A1.
| Описание полиморфизмов в экзонах | ||||||||
| ген | координата (chr13) |
референс | чтение | тип замены | синонимичность замены | глубина покрытия | качество чтений | аминокислотная замена |
| PHF11 | 50080847 | A | G | гомозиготная | да | 45 | 221.999 | L18L, L57L |
| COL4A1 | 110804809 | G | А | гетерозиготная | да | 26 | 166.009 | S1600S |
| COL4A1 | 110813709 | G | A | гетерозиготная | да | 46 | 166.009 | A1490A |
| COL4A1 | 110818598 | T | G | гетерозиготная | нет | 49 | 212.009 | Q1334H |
| COL4A1 | 110827684 | C | A | гетерозиготная | нет | 27 | 15.1417 | V1027L |
| COL4A1 | 110839550 | T | G | гомозиготная | нет | 24 | 221.999 | T555P |
| COL4A1 | 110864225 | A | T | гомозиготная | да | 47 | 221.999 | A144A |
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr13.dbsnp -build hg19 -dbtype snp138 snp.avinput
/nfs/srv/databases/annovar/humandb/
Полученнные файлы:
Судя по данным в файлах, 109 полиморфизмов имеют rs и 15 - неаннотированы. Гены без rs демонствируют в целом худшее качество и уровень покрытия.
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out
chr13.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
Тип аннотации: region-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr13.gwas
-build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
| Гены, имеющее клинические значение | |
| Координата | Значение |
| 25533831 | Obesity-related traits (Признаки ожирения) |
| 50080847 | Cardiac hypertrophy (Гипертрофия сердца) |
| 110818598 | Arterial stiffness (негибкость суставов) |
Тип аннотации: filter-based annotation.
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out
chr13.clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
| № | Команда | Назначение |
|---|---|---|
| 1 | fastqc chr13.fastq | Анализ качества чтений исходной последовательности |
| 2 | java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr13.fastq chr13_trim.fastq TRAILING:20 MINLEN:50 | Очистка чтений |
| 3 | fastqc chr13_trim.fastq | Анализ качества чтений очищенной последовательности |
| 4 | bwa index chr13.fasta | Индексирование референсной последовательности |
| 5 | bwa mem chr13.fasta chr13_trim.fastq > chr13.sam | Выравнивание очищенных чтений с референсной последовательностью |
| 6 | samtools view chr13.sam -b -o chr13.bam | Перевод выравнивания в бинарный формат .bam |
| 7 | samtools sort chr13.bam -T temp.txt -o chr13_sort.bam | Сортировка выравнивания чтений с референсом по координате в референсе начала чтения |
| 8 | samtools index chr13_sort.bam | Индексирование отсортированного выравнивания |
| 9 | samtools idxstats chr13_sort.bam > results.out | Выяснение числа чтений, откартированных на геном |
| 12 | samtools mpileup -uf chr13.fasta chr13_sort.bam -o snp.bcf | Создание файла с полиморфизмами в формате .bcf |
| 13 | bcftools call -cv snp.bcf -o snp.vcf | Создание файла со списком отличий между референсом и чтениями в формате .vcf |
| 14 | perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_mod.vcf -outfile snp.avinput | Подготовка входного файла для ANNOVAR |
| 15 | perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr13.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Refgene |
| 16 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr13.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Dbsnp |
| 17 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr13.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в 1000 genomes |
| 18 | perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr13.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Gwas |
| 19 | perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr13.clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация в Clinvar |
© Кучеренко Варвара 2015