Предсказание вторичной структуры тРНК (pdb id 1n78)
Предсказание вторичной структуры тРНК путем поиска инвертированных повторов и по алгоритму Зукера.
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях
олучает на вход последовательность нуклеотидов, а на выходе дает файлы sequence.fasta и sequence.inv, содержащие информацию
об обнаруженных комплементарных участках последовательности и предполагаемых водородных связях на этих участках соответственно).
Найдем возможные комплементарные участки в последовательности исследуемой тРНК и сравним с их описанием, полученным ранее с помощью find_pair.
После запуска программы с параметрами по умолчанию, файл sequence.fasta оказывался пустым, поэтому для корректной работы пришлось подбирать параметры вручную.
Наиболее резултативным оказалось изменение параметра Minimum score threshold, который по умолчанию был равен 50.
Программа выдала результат при понижении threshold до 16. При дальнейшем уменьшении параметра выходные данные не изменялись.
В конечном итоге удалось получить более-менее близкое к истине предсказание структуры только акцепторного стебля (5 пар из 7).
Следующая программа, предсказывающая вторичную структуру тРНК - RNAfold из пакета Viena Rna Package, использующая алгоритм Зукера.
RNAfold принимает на вход нуклеотидную последовательность и рассчитывает вторичную структуру РНК с минимальной свободной энергией (mfe)
в формате DBN (Dot-Bracket Notation).
Точки обозначают неспаренные нуклеотиды, круглые скобки - спаренные. Квадратные и фигурные скобки обозначают взаимодействия, формирующие псевдоузлы.
RNAfold полностью верно предсказала структуру акцепторного стебля и достаточно точно определила структуру Т- и D- стеблей.
Результаты занесены в таблицу ниже |
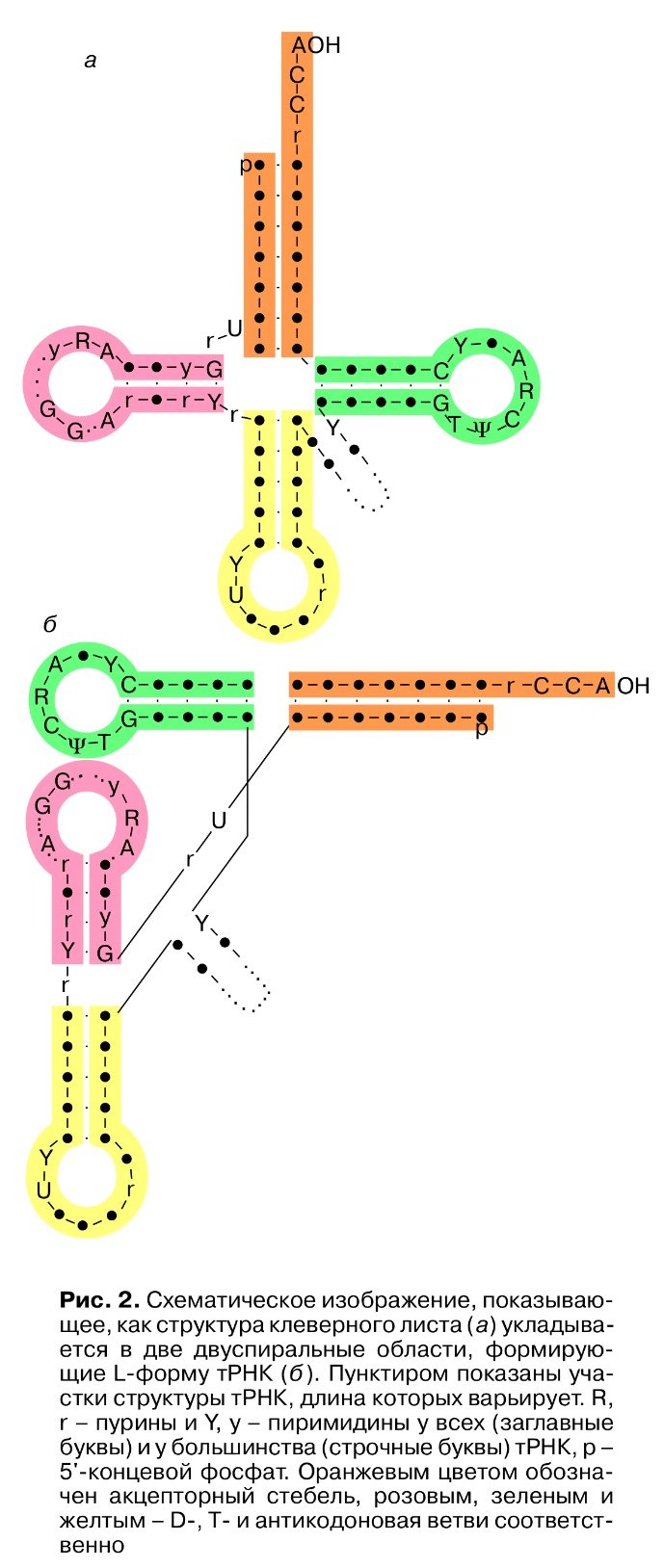
Рис.2[1] |
Реальная и предсказанная вторичная структура тРНК из файла 1n78.pdb
| Участок структуры | Позиции в структуре
(по результатам find_pair) | Результаты предсказания
с помощью einverted | Результаты предсказания
по алгоритму Зукера |
| Акцепторный стебель | 5'-501-507-3'
3'-572-566-5'
Всего 7 пар | 5'-3-7-3'
3'-69-65-5'
Всего 5 пар (совпдают все, но 2 пар не хватает) | 5'-501-507-3'
3'-571-565-5'
Всего 7 пар (все совпадают) |
| D-стебель | 5'-510-513-3'
3'-525-522-5'
Всего 4 пары | - | 5'-9-13-3'
3'-26-22-5'
Всего 5 пар (4 пары совпадают, одна лишняя) |
| T-стебель | 5'-549-553-3'
3'-565-561-5'
Всего 5 пар | - | 5'-48-52-3'
3'-63-59-5'
Всего 5 пар (сдвиг: 4 пары совпадают) |
| Антикодоновый стебель | 5'-538-544-3'
3'-532-526-5'
Всего 7 пар | - | 5'-27-31-3'
3'-39-43-5'
Всего 5 пар (все найденные пары совпадают, но 2 пар не хватает) |
| Общее число канонических пар нуклеотидов | 23 | 5 | 22 |
Исходя из данных таблицы, можно сделать однозначный вывод, что для данной структуры использование алгоритма Зукера оказалось более эффективным способом определения вторичной структуры.
| Предсказанная вторичная структура тРНК с минимальной свободной энергией, RNAfold |
Точечный график, показывающий вероятность образования пар оснований, RNAfold |
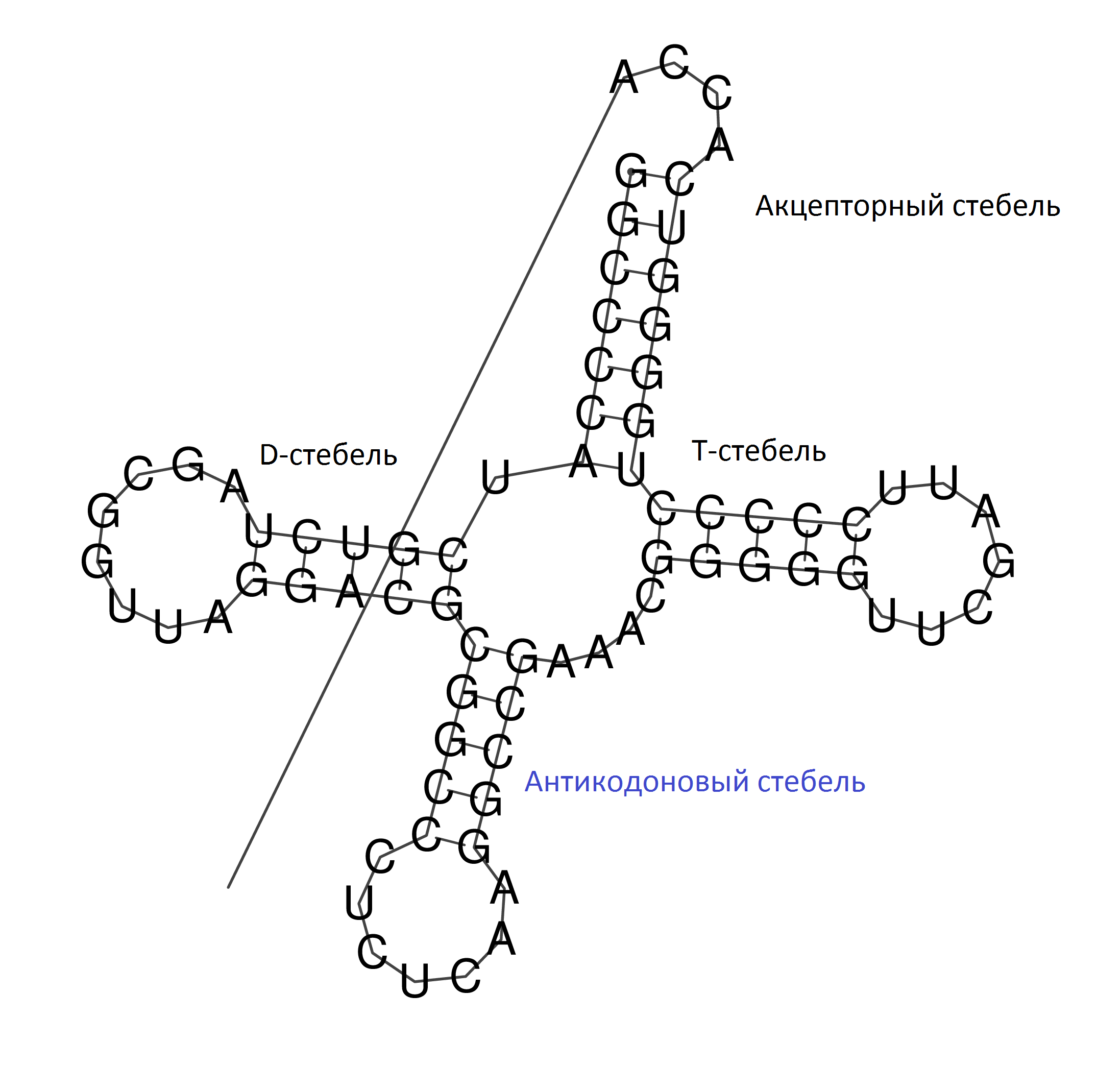 | 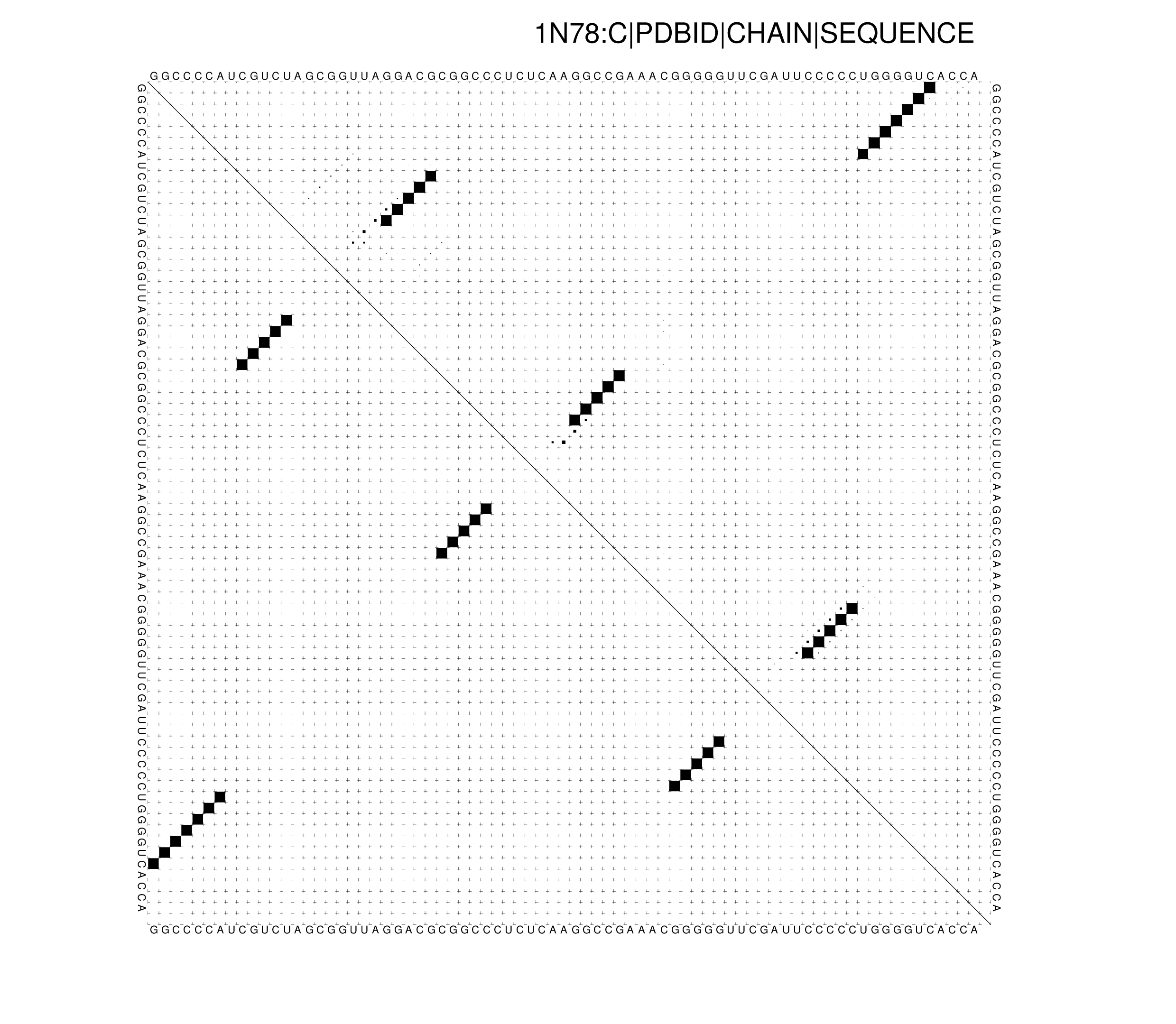 |
Поиск ДНК-белковых контактов (pdb id 1i3j)
С помощью команды define JMol были заданы множества атомов кислорода 2'-дезоксирибозы (set1), кислорода в остатке фосфорной кислоты (set2), азота в азотистых основаниях (set3)
(ссылка на скрипт)
Далее были найдены ДНК-белковые контакты (ссылка на скрипт).
Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5Å.
Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5Å.
Результаты приведены в таблице
Контакты атомов белка с:
|
полярные
|
неполярные
|
всего
|
Остатками 2’-дезоксирибозы
|
7
|
46
|
53
|
Остатками фосфорной кислоты
|
15
|
14
|
29
|
Остатками азотистых оснований со стороны большой бороздки
|
0
|
5
|
5
|
Остатками азотистых оснований со стороны малой бороздки
|
1
|
9
|
10
|
Из результатов видно, что наибольшее количество контактов в конмплесе - между белком
и остатками 2'-дезоксирибозы (большинство неполярных). Наименьшее количество контактов - между
белком и остатками азотистых оснований со стороны большой бороздки. Из этого можно сделать вывод,
что белок сильнее всего взаимодействует с остовом ДНК. Возможно, в связи с пространственной структурой белка.
Получение популярной схемы ДНК-белковых контактов с помощью программы nucplot
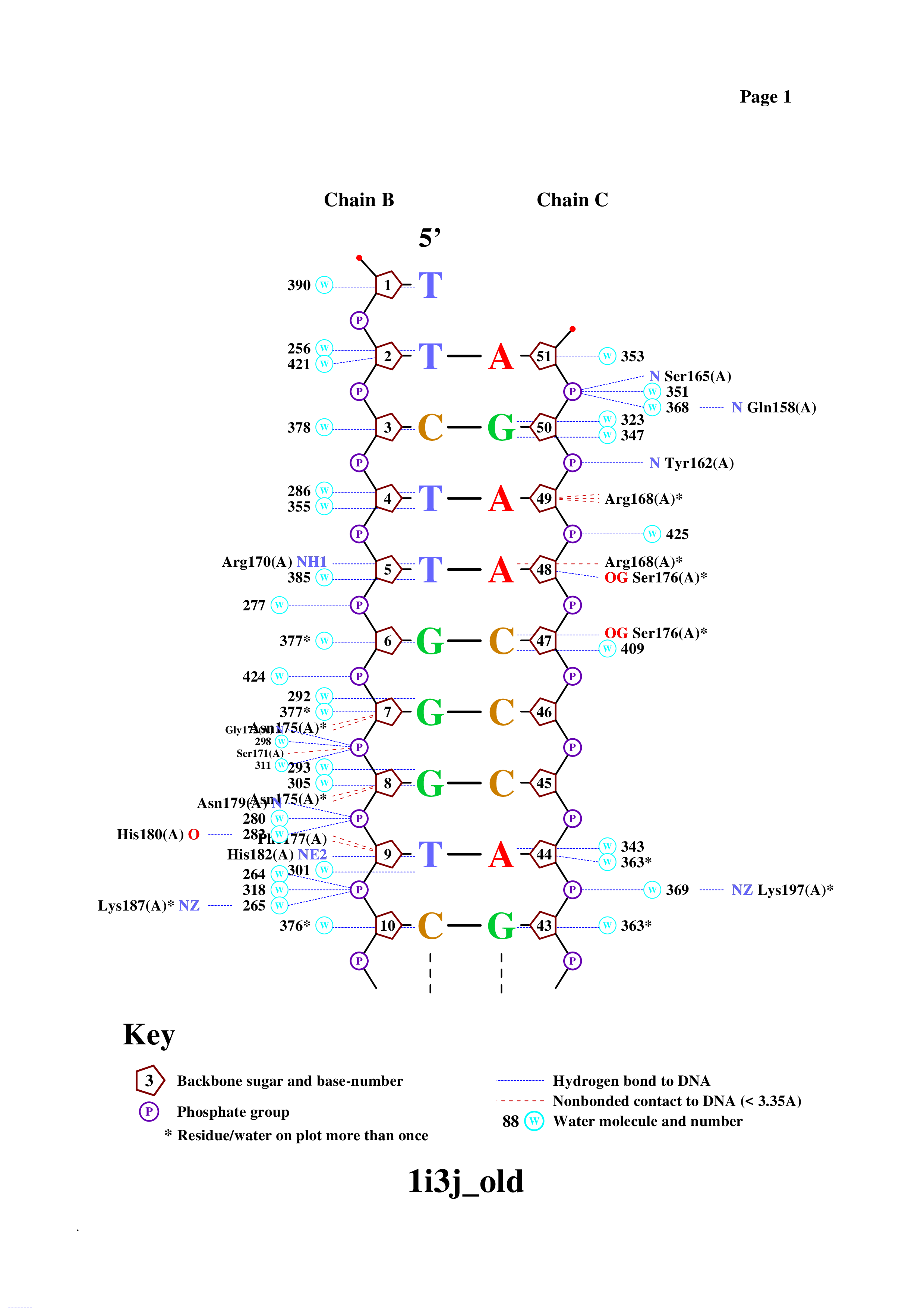
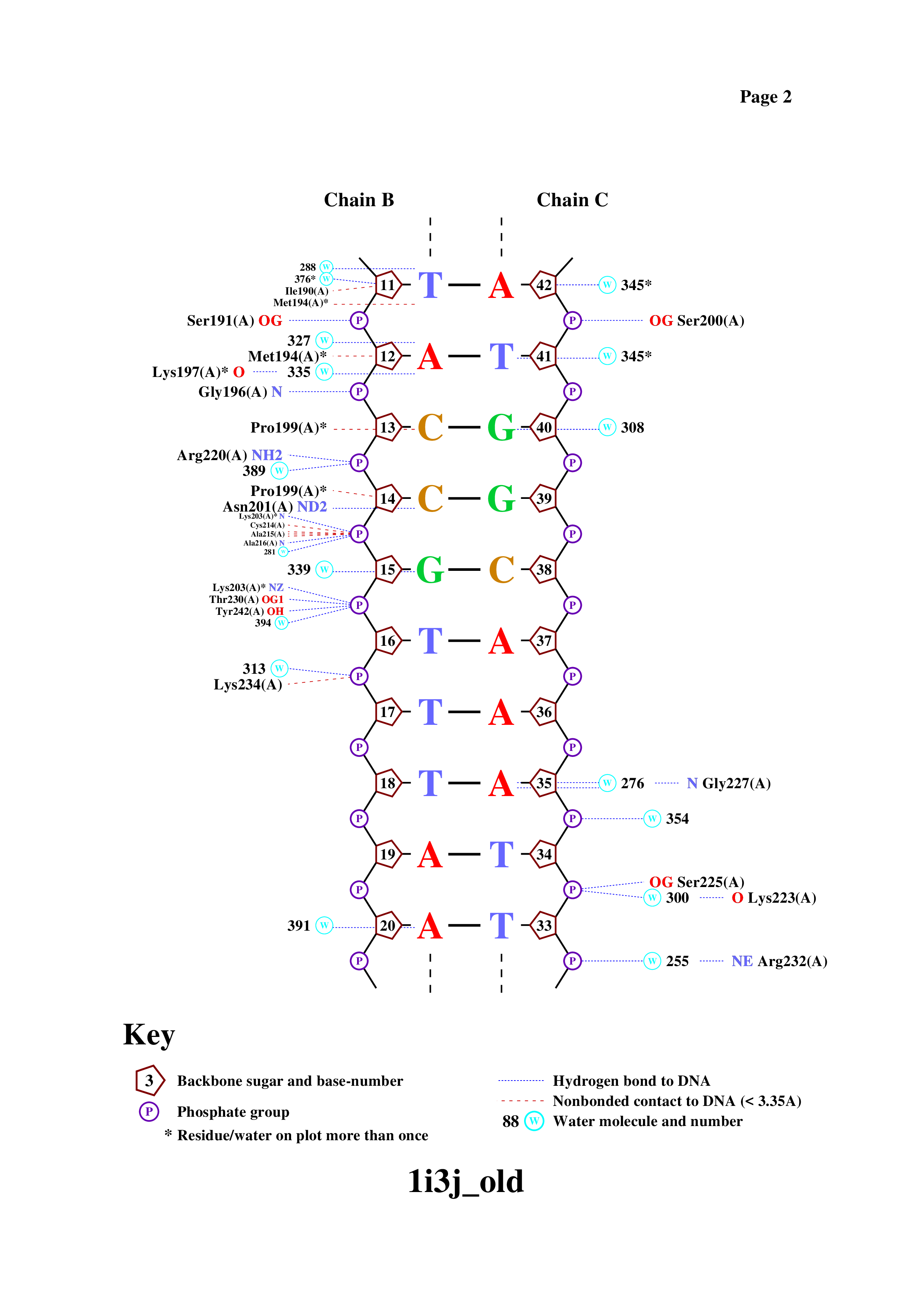
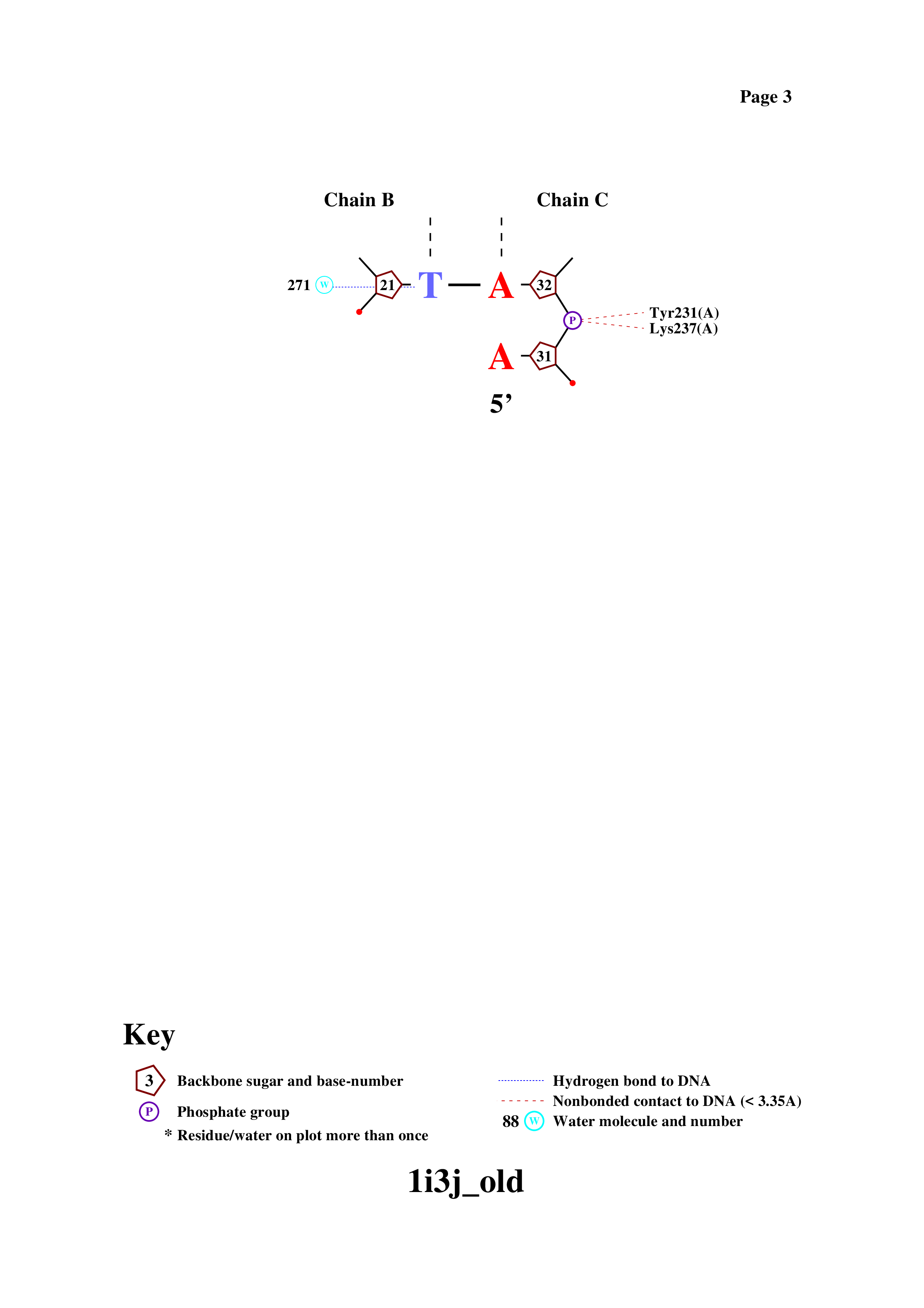
Аминокислотный остаток с наибольшим числом контактов с ДНК указанных на схеме - это Arg168(A) (4 контакта) и Asn175(A) (4 контакта).
Я думаю, что самые важные остатки для распознования последовательности ДНК -
это Arg168(A) (1 контакт с A48 цепи С), Met194(A) (1 контакт с Т11 цепи В) и Pro199(A) (1 контакт с С13 цепи В).
Предположительно контакты аминокислотных остатков с остатками азотистых оснований наиболее существенны,
так как они обеспечивают специфическое связывание и таким образом дают возможность распознать последовательности ДНК.
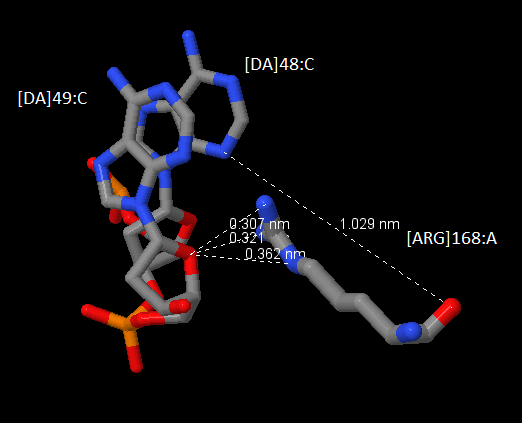
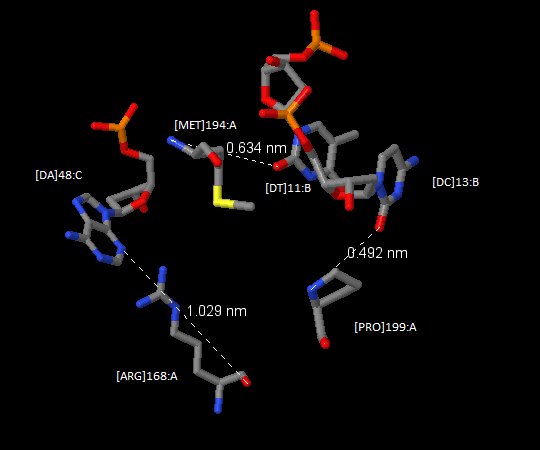
Ниже представлены изображения, полученные с помощью программы Jmol,
иллюстрирующие контакты выбранных остатков с ДНК.
 |
 |
| Аминокислотный остаток Arg168(A) с 4 контактами с ДНК: 3 контакта с [DA]49:C и 1 с [DA]48:C |
Контакты Arg168(A), Met194(A) и Pro199(A) c остаками азотистых основанийc [DA]48:С, [DT]11:B и [DC]13:B |
Источники
Назад
На главную
© Кучеренко Варвара 2015