Сборка de novo
Требовалось использовать пакет velvet для сбора из всех прошедших очистку чтений полученного набора контиги без использования референса. Были применены следующие команды:
- velveth vel 31 -fastq trim3.fastq - для выделения из чтений k-меров длиной 31;
- velvetg vel - для сборки контигов на основе графа k-меров.
Результат - собрано 156 контигов, N50 - 266, максимальная длина контига - 15264:

Задание 2.
Требовалось сравнить получившиеся контиги с последовательностью хромосомы программой blastn (алгоритм megablast). Команды:
- makeblastdb -in chr16.fasta -dbtype nucl - создаёт базу данных из последовательности 11 хромосомы;
- blastn -db chr16.fasta -query vel/contigs.fa -outfmt 7 -out vel.out - выполняет сравнение получившихся контигов с последовательностью 16 хромосомы.
Файл vel.out был обработан в Excel. В этом файле на первом листе расположены все контиги, на второй - только те контиги, которые картировались на хромосому единственным образом - таких нашлось 73. Некоторые из них картировались на одно и то же место:
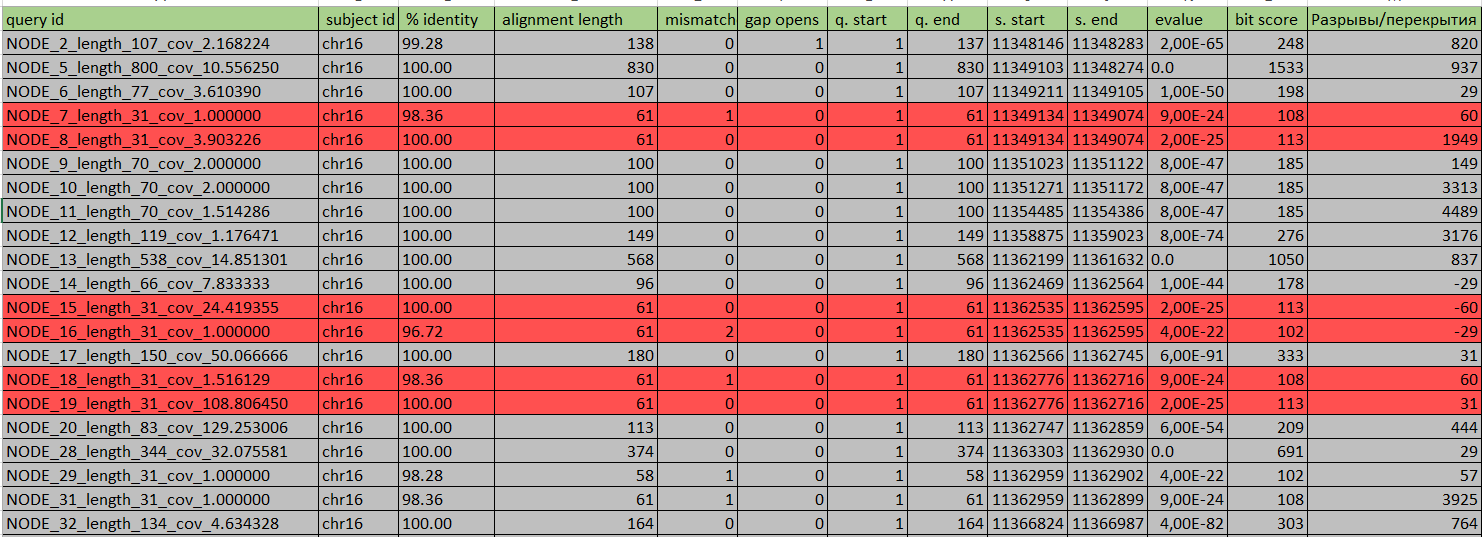
Охарактеризуем картированные единственным образом контиги:
- Много крупных разрывов между контигами (1000 - 5000 п.н.) - скорее всего, это интроны:
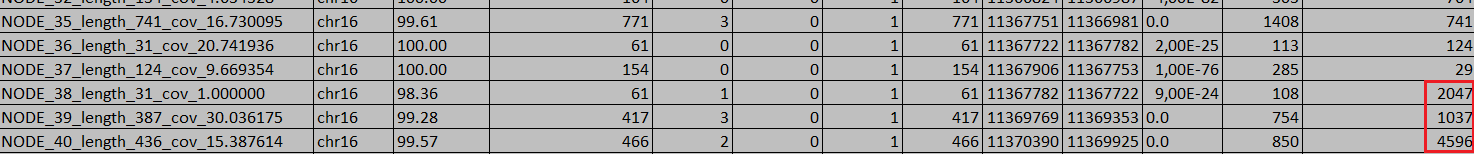
Рис.3. Пример гипотетического интрона. - Крупные разрывы в несколько сотен тысяч п.н. - межгенная область, т.к. это количество многократно превышает среднее значение длины интронов у человека:

Рис.4. Пример гипотетической межгенной области.