Эволюционные домены
Задание 1. Построение выравнивания представителей домена Pfam белков с разной доменной архитектурой
Для выполнения задания был выбран домен AAA_28 (ID PF13521) ААА-белков (ATPases
Associated with diverse cellular Activities).
Семейство ААА, или ААА+ включает в себя разнообразные по свойствам АТФазы, встречающиеся как у эукариот, так и у прокариот. Используют энергию гидролиза АТФ для
внесения изменений в структуру или свойства субстрата.
В том числе, вовлечены в процессы репликации ДНК, деградации белков, разрыва микротрубочек, передачи сигналов и регуляции экспрессии генов.
Домен AAA_28 содержит 671 белковую последовательность, принадлежащие 514 видам, 1 PDB-структуру и 18 белковых архитектур.
Выравнивание было импортировано в JalView из Pfam, раскрашено с помощью ClustalX, единственная известная PDB-структура - 1LW7 была связана со своей белковой последовательностью NADR_HAEIN и открыта в новом окне. jvp-проект доступен по ссылке.
Скрипт swisspfam_to_xls.py был запущен с параметрами -i /srv/databases/pfam/swisspfam.gz -z -p PF13521 -o aaa.xlsx. Результат доступен в Excel-таблице на вкладке "ааа".
Был составлен список последовательностей с указанием доменной архитектуры. Была создана сводная таблица, из которой были выбраны две доменные архитектуры: W8W0K1_9FLAO, содержащая домен AAA_28 и 574 последовательности, и K8WCN8_PRORE, содержащая домены AAA_28 и HTH_3 и 42 последовательности. Домен ААА_28 принадлежит к уже упомянутым АТФазам ААА+, а домен HTH_3 - это мотив спираль-поворот-спираль, состоящий из двух альфа-спиралей, соединенных коротким "мостиком". Часто встречается в белках, регулирующих экспрессию генов. Сводная таблица находится на вкладке "arch", выбранные домены выделены цветом.
Далее была получена информация о таксономической принадлежности: для этого из Uniprot был скачан файл, содержащий все последовательности всех белков из сводной таблицы, который после был обработан скриптом uniprot-to-taxonomy.py.
После того, как в таблицу была добавлена информация о доменной архитектуре и длине домена AAA_28 в каждой последовательности, был выбран таксон и два его подтаксона для сравнения. В качестве таксона я выбрал домен Bacteria, а в качестве подтаксонов - типы Actinobacteria и Proteobacteria. Эти подтаксоны были наилучшим образом представлены в архитектурах. К сожалению, тип Actinobacteria недостаточно хорошо представлен во второй доменной архитектуре.
Выбранные белки отмечены в сводной таблице буквой "Y".
Последовательность исследуемого домена из всех выбранных белков находится в новом jvp-проекте (ссылка). Последовательности разбиты на группы по принадлежности к одной из двух доменных архитектур.
Задание 2. Построение филогенетического дерева последовательностей домена.
Были введены следующие обозначения:
Приписав эти двубуквенные обозначения к началу последовательностей, с помощью программы MEGA методом maximum likelyhood было построено филогенетическое дерево:

На рисунке хорошо видно, что дерево разделяется на две клады, по доменной архитектуре. Это говорит о том, что видообразование внутри таксона Bacteria началось уже после того, как разошлись эти две доменные архитектуры. Под расхождением доменных архитектур я подразумеваю инсерцию домена HTH_3. Отсутствуют случаи вставки или исчезновения этого домена внутри клад. По моему мнению, это может говорить о сильно разнящейся функции белков из этих архитектур: как уже было упомянуто, домен HTH_3 характерен для белков, регулирующих генную экспрессию, в то время как исследуемый домен вовлечён в более широкий спектр процессов. Таким образом, двудоменная архитектура с большей вероятностью характерна для белков контроля экспрессии, в то время как для однодоменной архитектуры это, скорее всего, не так. Этот вывод находит подтверждение на дереве: в двудоменной архитектуре часто встречаются факторы регуляции транскрипции, а в однодоменной они отсутствуют.
Задание 3. HMM-профиль и построение ROC-кривой.
Для построения НММ-профиля было выбрано двудоменное семейство (см. Рисунок). Для получения HMM-профиля были предприняты следующие действия:
Результат работы этих трёх программ - HMM-профиль. С его помощью нужно было построить ROC-кривую. С выполнением этого задания для моего домена возникли проблемы, поэтому в качестве исходных данных для построения ROC-кривой была взята таблица lim.xlsx с диска P. Все вычисления приведены в таблице ROC.xlsx. Сначала было подсчитано общее количество положительных (31) и отрицательных (2043) вхождений исследуемых белков в lim-семейство (колонка №2). Затем, с помощью функции СЧЁТЕСЛИ, было подсчитано количество истинно положительных и истинно отрицательных результатов для каждого значения Nscore (колонки TP и TN). Разделив каждое из получившихся значений на общее количество соответствующих находок, я получил информацию для построения ROC-графика:
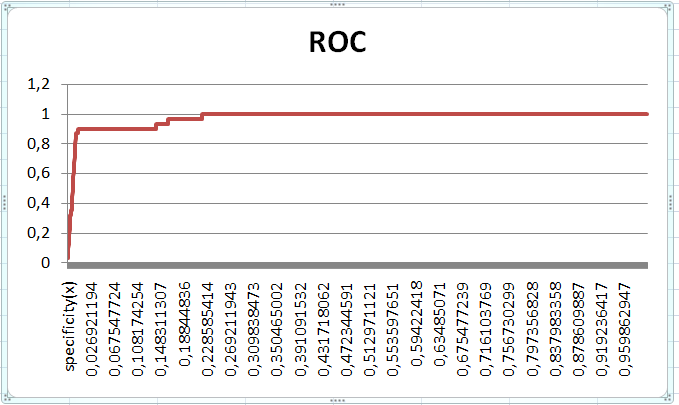
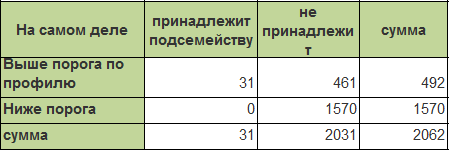
При визуальном анализе получившейся кривой был найден порог Nscore, при котором будут достигнуты максимальные специфичность и чувствительность. Nscore=14,631:
Назад к странице семестров