Молекулярная динамика биологических молекул в GROMACS:
Моделирование самосборки липидного бислоя и анализ результатов
Для получения исходной структуры набора липидов на kodomo были выполнены следующие команды:
# получим файлы
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/dppc.itp
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/lipid.itp
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/dppc.gro
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/b.top
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/em.mdp
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/pr.mdp
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/md.mdp
# создадим ячейку
genconf -f dppc.gro -o b_64.gro -nbox 4 4 4
# c помощью editconf преобразуем dppc.gro и b_64.gro в pdb файлы
editconf -f b_64.gro -o b_64.pdb
editconf -f dppc.gro -o dppc.pdb
# В текстовом редакторе в файле b.top установили правильное количество липидов в системе
mcedit b.top
# Сделаем небольшой отступ в ячейке от липидов, что бы добавить примерно 2500 молекул воды
editconf -f b_64.gro -o b_ec -d 0.5
# Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты молекул
grompp -f em -c b_ec -p b -o b_em -maxwarn 2
mdrun -deffnm b_em -v
# Cтоит отметить изменение максимальной силы в ходе оптимизации геометрии:
# от 4.37970е+05 на шаге 0 до 6.16887е+02 на шаге 51
# Добавим в ячейку молекулы воды типа spc
genbox -cp b_em -p b -cs spc216 -o b_s
# Проведём "утряску" воды:
grompp -f pr -c b_s -p b -o b_pr -maxwarn 1
mdrun -deffnm b_pr -v
# Переформатируем b_pr.gro и b_s.gro в pdb формат
editconf -f b_pr.gro -o b_pr.pdb
editconf -f b_s.gro -o b_s.pdb
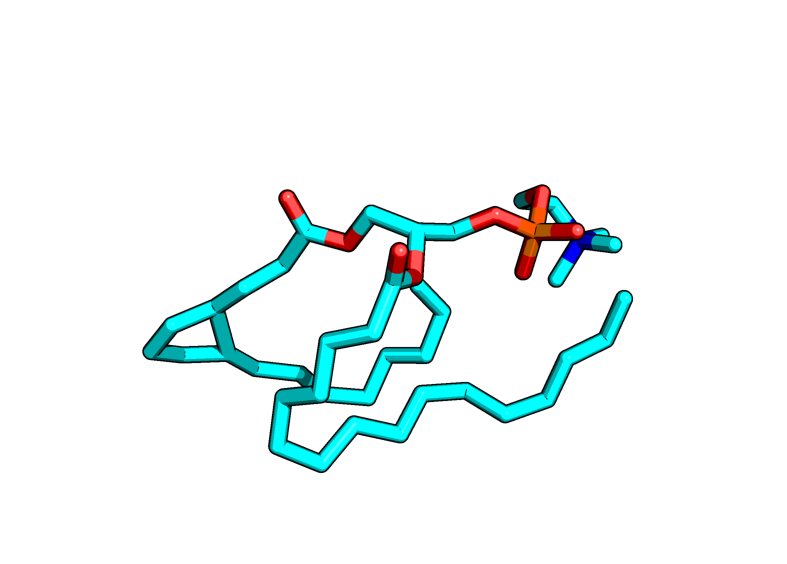 |
 |
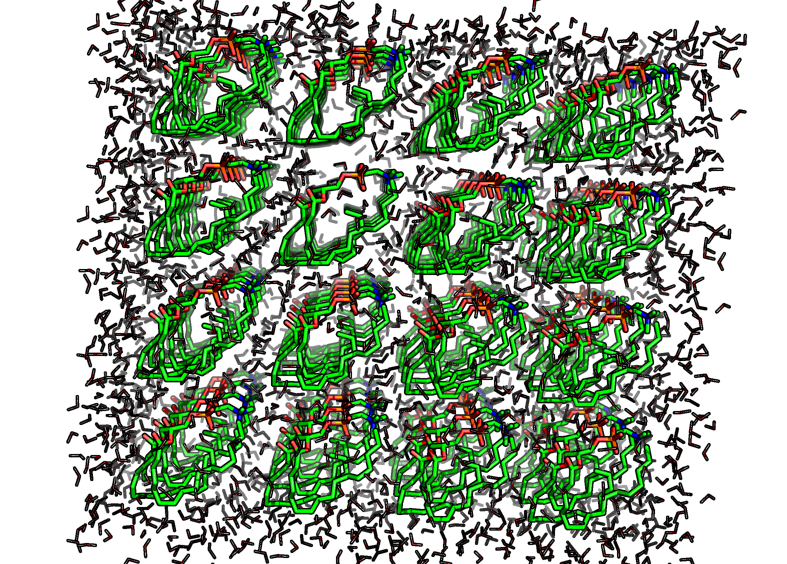 |
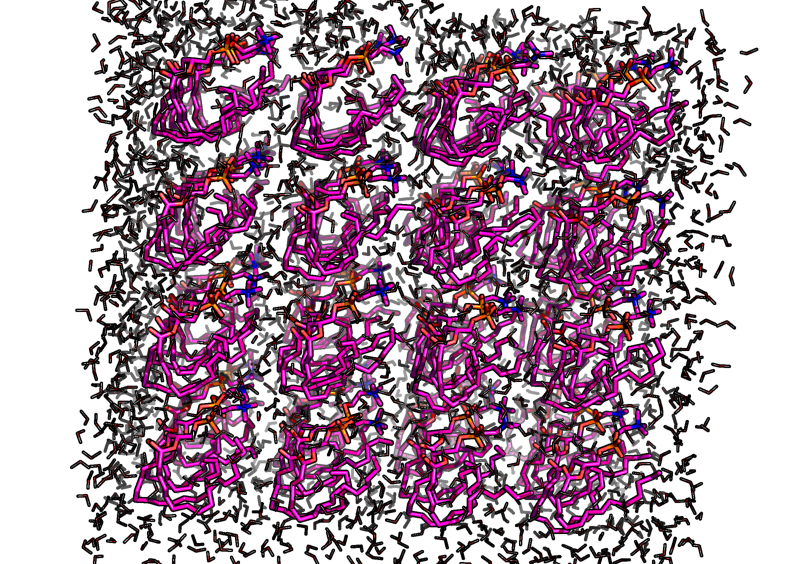 |
Видим, что после утряски липидный параллелипипед стал более расхлябанным, гидрофобные хвосты теперь висят как попало.
sbatch -N1 --ntasks-per-node=2 -e error-gpu.log -o output.log -t 350 -p gpu impi /opt/ccoe/gromacs-5.0.4/build/bin/gmx_mpi mdrun -testverlet -deffnm b_md -v
Анализ результатов моделирование самосборки липидного бислоя
# Любой анализ начинают с визуального анализа движений молекул. При вопросе о выводк групп выберите DPPC
trjconv -f b_md.xtc -s b_md.tpr -o b_pbc_1.pdb -skip 20
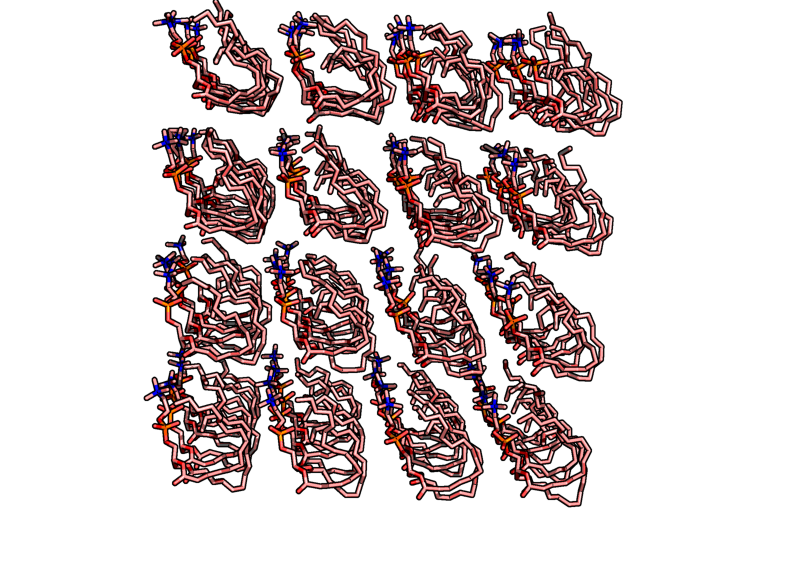 |
 |
Попробуем выставить Periodic Boundary Conditions при помощи опции -pbc mol:
trjconv -f b_md.xtc -s b_md.tpr -o b_pbc_2.pdb -skip 20 -pbc mol

Система организуется в мицеллоподобную структуру с самого начала моделирования, однако нечно похожее на бислой наблюдается на начиная с фрейма 35. Из pdb файла найдем время образования этой модели: 17000.00

Определим площадь занимаемую одним липидом.
import pandas as pd
# размеры ячейки из траектории
g_traj -f b_md.xtc -s b_md.tpr -ob box_1.xvg
with open('box_1.xvg', 'r') as f:
lines = [item.strip('\n') for item in f.readlines()
if (not item.startswith("@")) and (not item.startswith("#"))]
times = [item.split('\t')[1] for item in lines]
sq = [float(item.split('\t')[3]) * float(item.split('\t')[4]) / 32 for item in lines]
data = pd.DataFrame({'times':times, 'Squares':sq})
data.head()
data.plot(x="times", y = "Squares")
plt.show()

Видим, что площадь, занимаемая средним липидом, с течением времени уменьшается за счет того, что липиды компактизуются в пространстве и укладываются в бислой. Даже испортивший нам всю картину кусок тоже относительно хорошо собран для одного слоя.
Теперь пределим изменение гидрофобной и гидрофильной поверхности в ходе самосборки:
g_sas -f b_md.xtc -s b_md.tpr -o sas_b.xvg
Нормаль к поверхность это ось Х
with open("sas_b.xvg", 'r') as f:
lines = [item.strip('\n') for item in f.readlines()
if (not item.startswith("@")) and (not item.startswith("#"))]
times = [int(item.split()[0]) for item in lines]
hphob = [float(item.split()[1]) for item in lines]
hphil = [float(item.split()[2]) for item in lines]
data = pd.DataFrame({'times':times, 'Hydrophobic':hphob, 'Hydrophilic':hphil})
data.head()
data.plot(x="times", y = ["Hydrophobic", "Hydrophilic"])
plt.show()

Вначале гидрофобная поверхность системы максимально: липиды лежат в ячейке отдельно, при этом стоит помнить и про возникшую при утряске системы с водой "расхлябанность". Однако это состояние крайне невыгодно, и система очень быстро (уже на втором фрейме) начинает скатываться в мицеллоподобную структуру, а затем утрясывается до бислоя, при этом гидрофобная поверхнсть продолжает уменьшаться (однако выитый наружу кусок немного ее увеличивает к концу). Гидрофильная повперхность всё это время выше, но она тоже несколько снижается из-за компактизации липидов в бислое.
Теперь оценим меру порядка в начале и в конце траектории:
wget http://kodomo.cmm.msu.ru/~golovin/bilayer/sn1.ndx
# начало
g_order -s b_md -f b_md.xtc -o ord_start.xvg -n sn1.ndx -e 5000 -d X
# конец
g_order -s b_md -f b_md.xtc -o ord_end.xvg -n sn1.ndx -b 45000 -d X
with open("ord_start.xvg", 'r') as f:
lines = [item.strip('\n') for item in f.readlines()
if (not item.startswith("@")) and (not item.startswith("#"))]
xax = [int(item.split()[0]) for item in lines]
start = [float(item.split()[2]) for item in lines]
with open("ord_end.xvg", 'r') as f:
lines = [item.strip('\n') for item in f.readlines()
if (not item.startswith("@")) and (not item.startswith("#"))]
end = [float(item.split()[2]) for item in lines]
data = data = pd.DataFrame({'xax':xax, 'Start_order':start, 'End_order':end})
data.head()
data.plot(x="xax", y = ["Start_order", "End_order"])
plt.show()

В начале траектории значения меры порядка сильно колебаются, что можно объяснить с хаотичностью движений молекул липидов. В конце траектории видно, что сначала липиды довольно сильно теряют подвижность в силу организации их в бислой, однако потом выбивается тот самый кусок, испортившй нам картину, и подвижность тут же резко возрастает.
Основные сведения о системе
- Силовое поле используемое при построении топологии топологии - ffgmx
- Заряд системы - 0.000 (всё протонировано?)
- Размер и формa ячейки - прямоугольный параллелепипед со сторонами 6.2600, 4.4430 и 5.7780 нм.
- Минимизация энергии:
- Модель, которой описывался растворитель - flexspc
- Утряска растворителя:
- Основной расчёт МД:
- Алгоритм минимизации энергии - l-bfgs
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - Cut-off
- Число шагов - 1000
- Длина шага - 0.0002
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - pme, Cut-off
- Алгоритмы термостата и баростата - Berendsen, no
- Время моделирования - 50000.00
- Длина траектории - 50 ns
- Число шагов - 10000000
- Длина шага - 0,005 ps
- Алгоритм интегратора - md
- Алгоритмы расчёта электростатики и Ван-дер-Ваальсовых взаимодействий - pme, Cut-off
- Алгоритмы термостата и баростата - v-rescale, Berendsen