Хемоинформатика
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit import RDConfig
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
import numpy as np
from IPython.display import display,Image
import rdkit.Chem.Lipinski as Lipinksy
В этой работе мы будем модифицировать молекулу ибопрофена имитируя реакции Click-химии, а затем отберем те из соединений, что удовлетворяют правилу пяти Лепински. Для начала визуализируем молекулу ибупрофена:
def drawSmilesStructure(string):
mol = Chem.MolFromSmiles(string)
AllChem.Compute2DCoords(mol)
display(mol)
ibu = 'CC(C)CC1=CC=C(C=C1)C(C)C(=O)O'
drawSmilesStructure(ibu)

Отлично. Теперь проверим, удовлетворяет ли ибупрофен правилу пяти Лепински:
def checkLepinsky(mol):
return bool((Lipinksy.NumHDonors(mol) <= 5) and (Lipinksy.NumHAcceptors(mol) <= 10) \
and (Lipinksy.rdMolDescriptors.CalcExactMolWt(mol) < 500) \
and(Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(mol)[0]) <= 5)
ibu_mol = Chem.MolFromSmiles(ibu)
AllChem.Compute2DCoords(ibu_mol)
checkLepinsky(ibu_mol)
Теперь перепишем формулу ибупрофена так, чтобы получить шаблон для замены азидного радикала, имитируя реакцию Click-химии:
template = 'N1N=NC(=C1)C(C1=CC=C(C=C1)CC(C)C)(C(=O)O)'
drawSmilesStructure(template)

В базе PubChem были найдены и сохранены в файл SMILES-структуры 141959 веществ, содержащих азид-радикал не в составе цикла и потенциально пригодных для проведения нашей реакции. Для начала отфильтруем вещества по размеру записи и наличию разрывов:
strings=np.genfromtxt('2515324818764782706.txt',dtype=np.str)
smiles = []
for line in strings:
if (len(line[1]) < 30) and (len(line[1]) > 11) and not ( '.' in line[1]):
smiles.append(line[1])
len(smiles)
products = []
# Новую молекулу лучше создавать в try из-за битых Smiles:
for smi in smiles:
try:
if not "N=[N+]=[N-]" in smi:
continue
newsmi = smi.replace("N=[N+]=[N-]", template)
if checkLepinsky(Chem.MolFromSmiles(newsmi)):
products.append(newsmi)
except Exception:
pass
len(products)
Посмотрим, что же у нас получилось:
from random import sample
sample_mols = [Chem.MolFromSmiles(item) for item in sample(products, 12)]
Chem.Draw.MolsToGridImage(sample_mols, molsPerRow=3, subImgSize=(500, 400))

Отличная выборка. Теперь построим similarity map исходной молекулы ибупрофена с, например, пятым веществом нашей выборки:
from rdkit.Chem.Draw import SimilarityMaps
ibumol = Chem.MolFromSmiles(ibu)
fig, maxweight = SimilarityMaps.GetSimilarityMapForFingerprint(ibumol, \
sample_mols[4], SimilarityMaps.GetMorganFingerprint)

maxweight
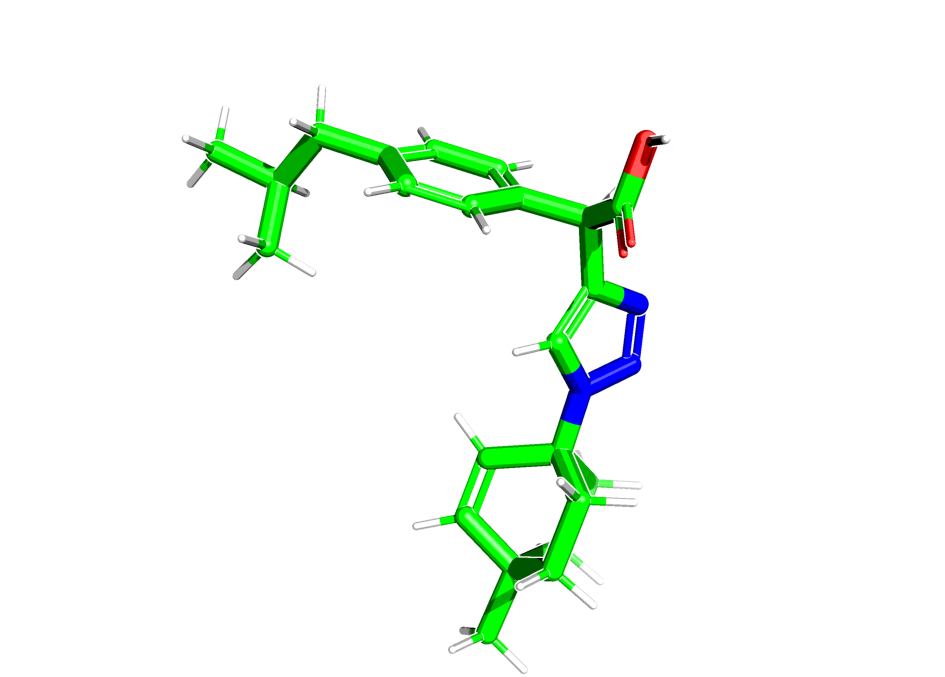
Теперь посмотрим на трехмерную структуру какой-нибудь молекулы из выборки, например 11-й:
def molTo3d(string):
m3d=Chem.AddHs(string)
Chem.AllChem.EmbedMolecule(m3d)
AllChem.MMFFOptimizeMolecule(m3d,maxIters=500,nonBondedThresh=200)
return m3d
randmol3d = molTo3d(sample_mols[10])
import nglview as nv
import ipywidgets
nv.show_rdkit(randmol3d)
Мне не удалось вылечить проблему с nglview, поэтому я построил .mol файл при помощи babel и открыл его в памоле:
Chem.MolToSmiles(randmol3d)
with open('smile.smi', 'w') as f:
f.write(Chem.MolToSmiles(randmol3d))
bash: obgen smile.smi > smile.mol
import __main__
__main__.pymol_argv = [ 'pymol', '-x' ]
import pymol
pymol.finish_launching()
from pymol import cmd, stored
cmd.load('smile.mol')
cmd.zoom()
cmd.turn("y", "90.0")
cmd.do('''
set antialias, 2
set ray_trace_mode, 3
''')
cmd.png(filename='pic1.png',width='8cm',dpi=300, ray=1)