Практикум 3
1. Сложные случаи для предсказания с помощью AlphaFold: амилоиды
Более-менее случайно решила взять последовательность A4. Если бегло взглянуть на молекулу, можно предположить, что она несёт скорее отрицательный заряд, и притом, я бы сказала, что включает довольно много ароматических аминокислотных остатков.
MGATAVSEWTEYKTADGKTFYYNNRTLESTW
Подаём на вход AlphaFold последовательность и предсказываем структуру отдельной молекулы, а также комплекса из 5 и 10 молекул, далее приведены лучшие структуры в выдаче. Полученные архивы выложены здесь. Нетрудно заметить, что получилось что-то, как минимум, странное.
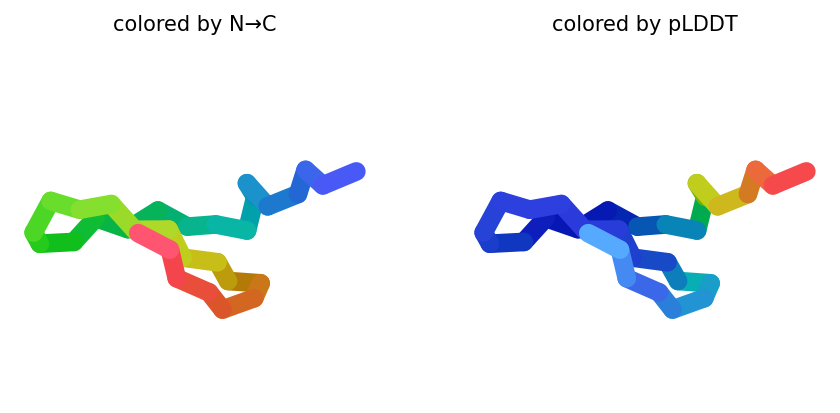
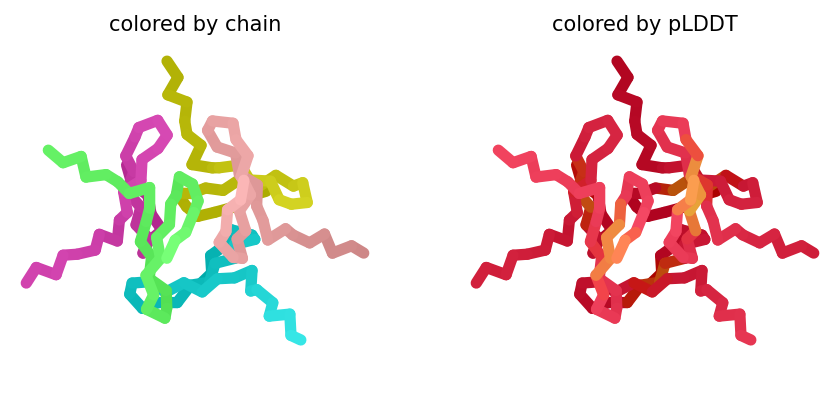
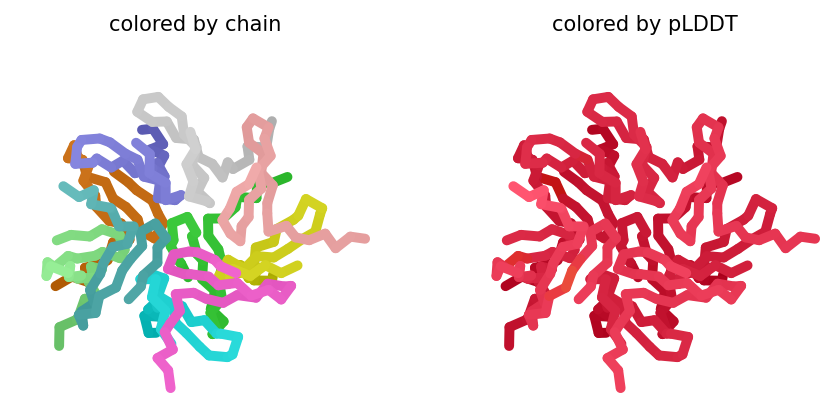
Я предполагаю, что AlphaFold сначала сворачивает белки, а потом пытается их как-то состыковать. Это можно проверить, просто наложив структуры друг на друга. Кроме того, проблема состоит в том, что он учился на экспериментальных структурных данных, а они, по большей части, описывают глобулярные белки, поэтому и на выходе видим что-то типа глобулярного комплекса, но кривенького, поэтому значения pLDDT не очень высокие. То есть модель сама понимает, что выдала какой-то бред, но как сделать правильно — она просто не знает.
Сравним с экспериментально полученной структурой (PDB ID 2NNT) - рис. 2, Б.
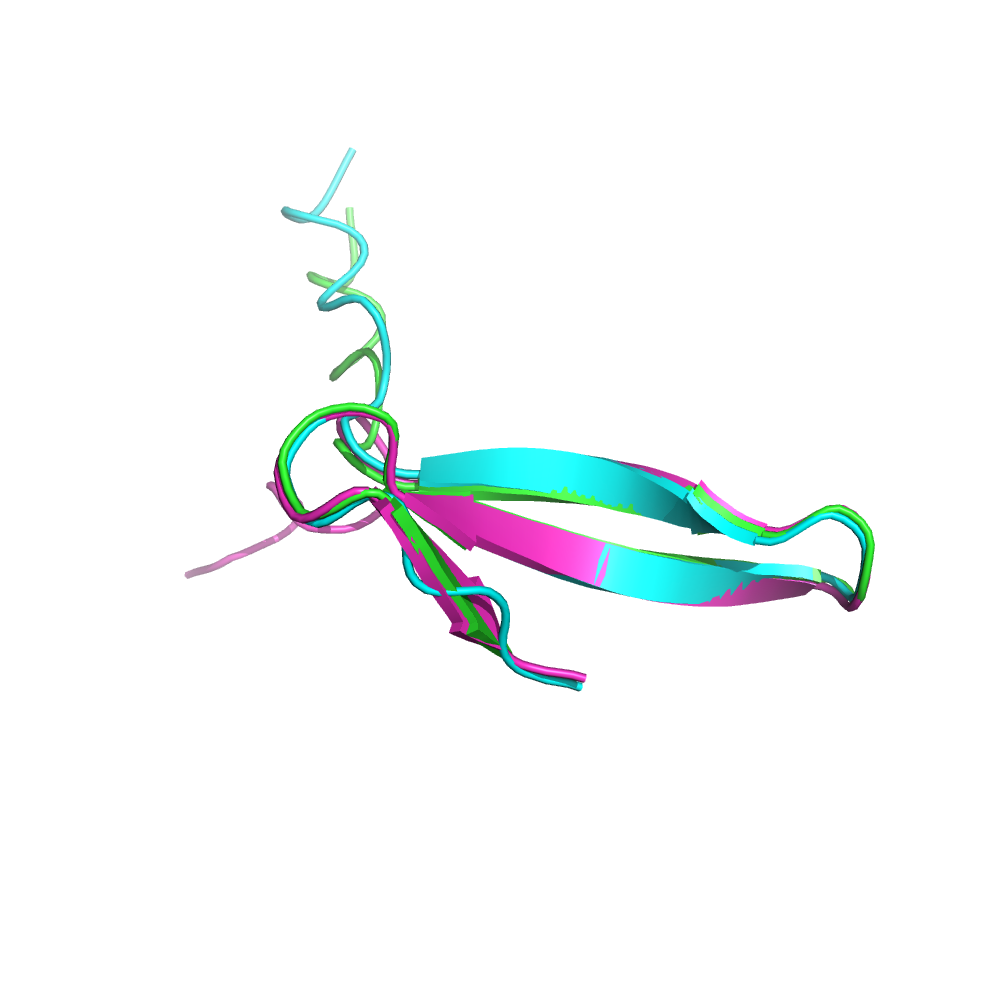
2. Разбиение на домены алгоритмом DOMAK
Дан белок (PDB ID 1IGT, chain B). Попробуем реализовать алгоритм DOMAK, который предполагает, что
внутри домена контактов больше, чем между ними. Сами контакты предсказали Arpeggio, после чего рассчитали
значения индекса разделённости для всех возможных позиций точки, которая разделяет два домена. Расчёты
приведены тут.
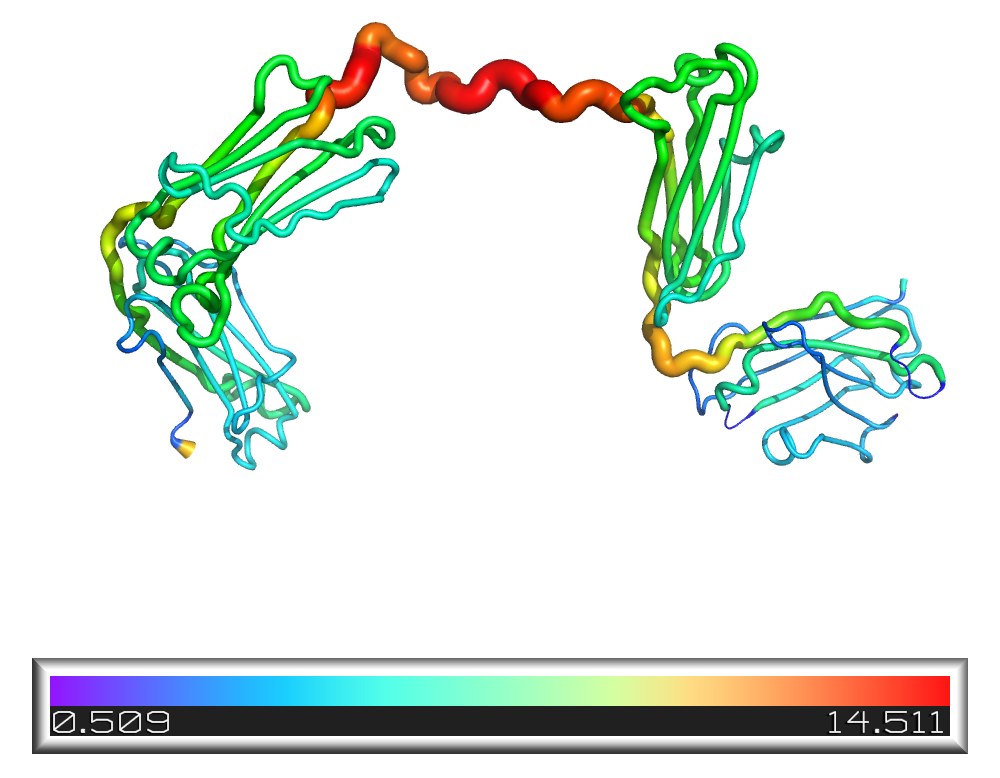
После я воспользовалась скриптом loadBfacts.py, чтобы после изобразить на структуре эти значения.
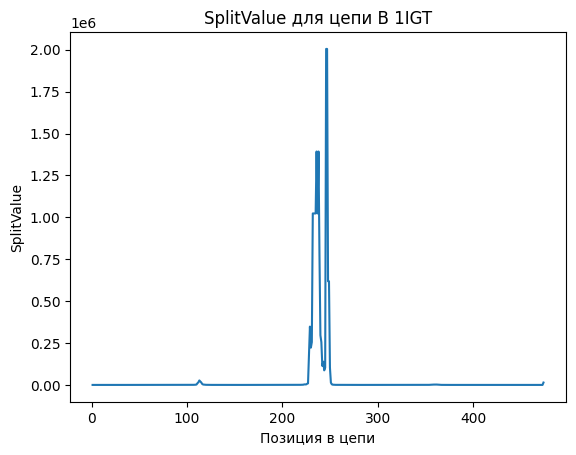
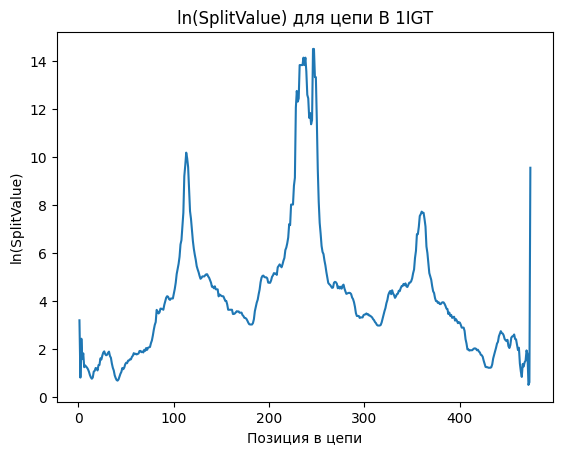
Правда, взглянув на структуру, я поняла, что, видимо, на том масштабе, который получился на графике
(рис. 4, А), видно не все элементы структуры. Первое, что мне пришло в голову — взять логарифм от этих
значений (рис. 4, Б). На этом графике вроде видно лучше, но лучше наложить на структуру (рис. 4, В), и тогда
становится видно, что в линкерах между доменами значение SplitValue правда выше. Кроме того, чем линкер длиннее,
тем более выраженный пик.
3. Сравнение с аннотацией доменов в базах данных
В PDB нашла аннотации доменов в базах данных SCOP и CATH. Если формально разбить последовательность на 4 домена по полученным выше пикам и сопоставить координаты, то получим следующее:
| Источник | Домен 1 | Домен 2 | Домен 3 | Домен 4 |
|---|---|---|---|---|
| SCOP | 1-119 | 120-223 | 224-342 | 343-444 |
| CATH | 1-119 | 120-224 | 238-341 | 342-444 |
| DOMAK | 1-112 | 113-245 | 246-359 | 360-474 |
С учётом того, что между доменами ещё есть какие-никакие линкеры, мне кажется, аннотации вполне соотносятся, хотя всё равно есть ощущение, что где-то съехала нумерация.