Практикум 12
Сравнение выравниваний, полученных с помощью разных программ
Для выполнения задания были взяты белки из практикума 9, п. 5: DNAA - белок, инициирующий хромосомную репликацию (DNAA_SERMA, DNAA_MYCTA, DNAA_TREPA, DNAA_STRCO, DNAA_YERPE). Для сравнения выравнивания одних и тех же последовательностей разными программами были выбраны алгоритмы Muscle (А), MAFFT (B), ClustalW (С). Программы были запущены с базовыми праметрами. Проект Jalview. С помощью сервиса VerAlign были найдены совпадающие блоки.
Muscle + MAFFT
- SP score: 0,96
- CS score: 0,94
- avg_SPdist score: 0,98
примеры несовпадающих блоков: (22,46)и(22,46), (111,126)и(111,126).
Muscle + ClustalW
- SP score: 0,92
- CS score: 0,90
- avg_SPdist score: 0,97
примеры несовпадающих блоков: (22,46)и(22,46), (53,54)и(53,54).
Первая пара содержит большие доли совпадающих позиций по цепи (SP) и по колонкам (CP), а также визуально имеет больше сопадающих блоков. Различия в выравниваниях объясняются тем, что программы работают по разным алгоритмам и по-разному разрешают участки с низкой степенью сходства. Наибольшим сходством обладают программы Muscle и MAFFT.
Построение выравнивания по совмещению структур и сравнение его с выравниванием MSA
Для выполнения задания было выбрано семейство ABC-транспортеров, которые отвечают за перемещение различных соединений через биологические мембраны (PF00005): PDB: 1GAJ, 1JI0, 1JJ7 (референс).
На основе попарного выравнивания структур на сервисе Pairwise Structure Alignment (метод TM-align) вручную было построено множественное выравнивание. Те же белки были выровнены алгоритмом Muscle. Проект Jalview.
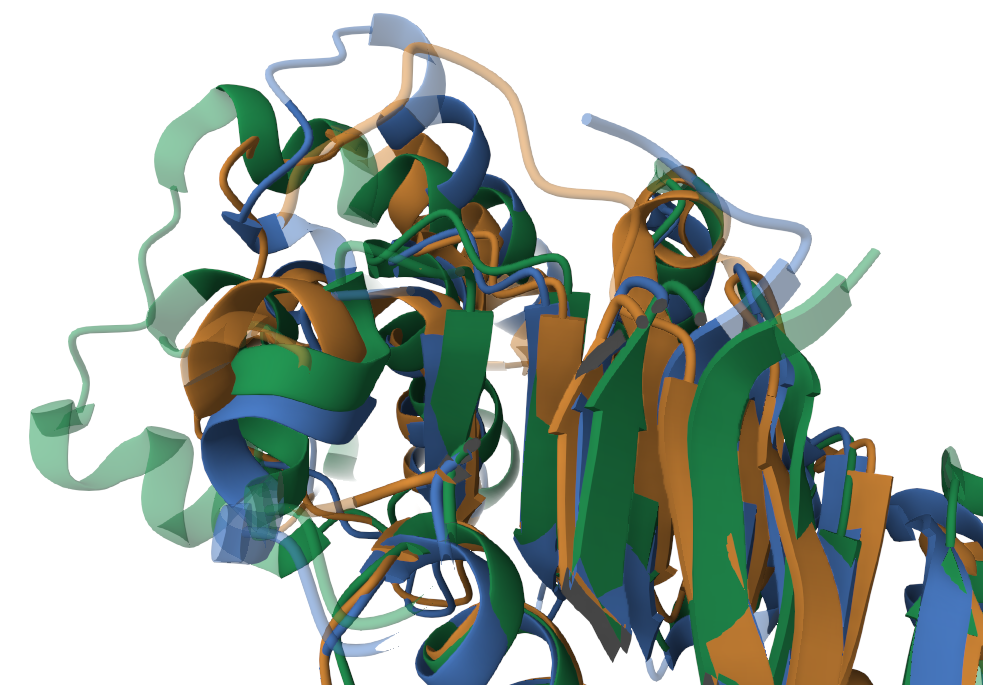
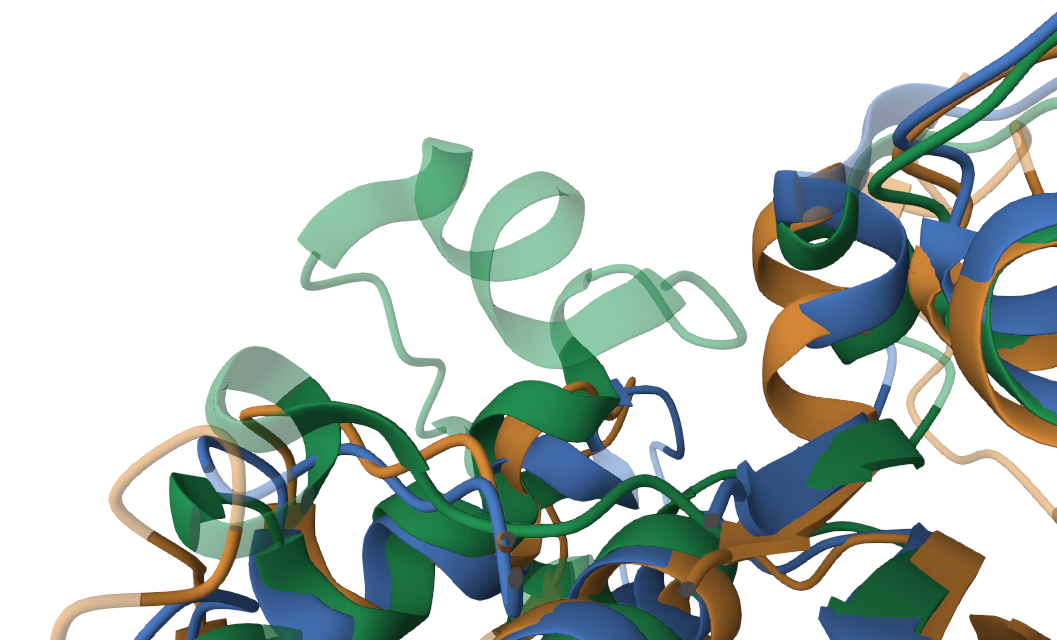
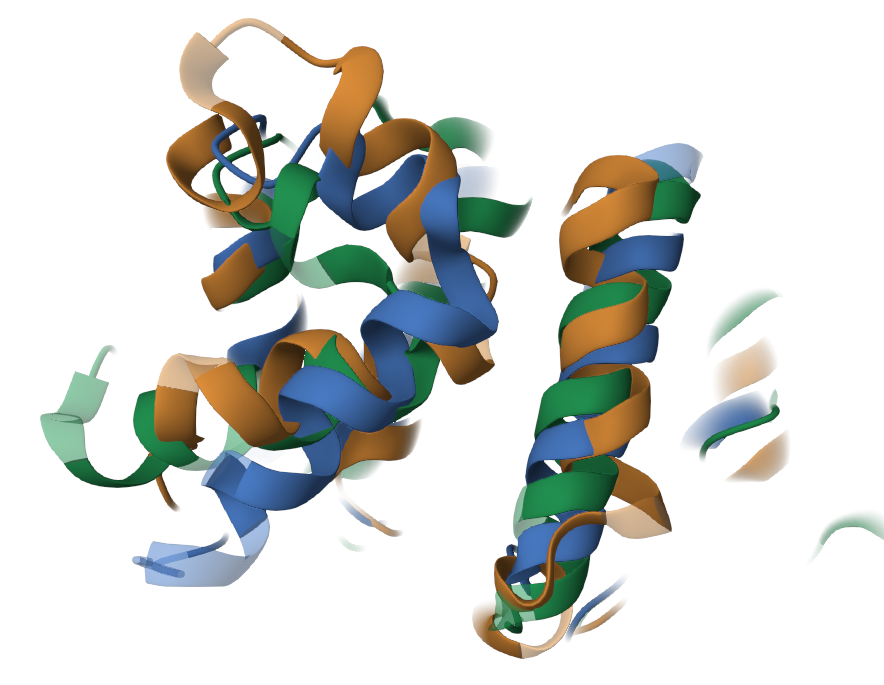
Данные белки состоят как из альфа-спиралей, так и из бета-листов. Стоит отметить, что невыровненными участками являются: (а) концевые участки (обусловлено различной длиной исследуемых белков, торчащие концы, рис.1), (б) вероятно дополнительная альфа-спираль, которой не с кем выровнятся (участок 120-140, рис.2), (в) предсказанные изгибы (повороты) альфа-спиралей (переходный участок 160-170, рис.3) .
Пространственное и програмное выравнивания обладают высоким сходством, имеют нескольок консервативных участков, из чего следует консервативность белков.
Описание команды Mafft
Mafft - программа множественного выравнивания (MSA), опубликованная в 2002 году. Аминокислотные последовательности задаются как векторы от компонента объема и компонента полярности, поскольку нейтральные мутации, затрагивающие данные параметры, сохраняют структуру белков, то есть последовательности с нейтральными мутациями могут быть выровнены. Вычисляется корреляция между двумя аминокислотными последовательностями (т.е. векторами) и строится попарное выравнивание. Используя рассчитанные попарные выравнивания, выполняется вычесление матрицы расстояний для оценки различий между выравниваниями. На основе матрицы расстояний строится направляющее дерево, в котором узлами являются кластеры, а ветви отражают расстояния между кластерами. По иерархии направляющего дерева выполняется прогрессивное выравнивание кластеров от листьев к корню. Последний этап - итеративное уточнение, повторяется весь процесс с корректировкой положения инделей для повышения точности выравнивания. MAFFT считается одним из наиболее точных и универсальных инструментов выравнивания нескольких последовательностей.
Источники: