BLAST
Работа со страничкой BLASTp
В начале предлагается указать, какую именно последовательность надо анализировать.
Есть возможность выбрать конкретный участок белка.
Далее надо уточнить, из какой базы данных требуется брать белки для выравнивания.
Опциональные возможности: указать конкретный организм, а также исключить записи refseq с определёнными идентификаторами.
Также можно выбрать алгоритм. На выбор даётся четыре:
1) Quick BLASTP - ускоренная версия BLASTP
Как это работает? (очень упрощённо)
1. найти кандидатов, не используя выравнивания (индексировать k-меры
из баз данных и введённой последовательньсти)
2. провести выравнивания с кандидатами из топ-1500 и упорядочить
выдачу, используя blastp
Такое ускорение даёт действительно внушительный выигрыш по времени.
Так, для того, чтобы проанализировать протеом Shigella flexneri SP1 WGS
используя BLASTP,
требуется около 10 часов, в то время как для QuickBLASTP это время составляет 0.8 часов.
Однако, надо держать в уме, что для identity, меньшем 65%[1], результаты Quick BLASPTP гораздо хуже,
чем таковые для BLASTP, что говорит о том, что ускоренный алгоритм следует использовать
для быстрого поиска очень близких белков.
2) blastp
3) PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool)
использует построение позиционой матрицы (длина х 20, ij-й элемент соответсвует
вероятности, что у близких последовательностей в позиции i стоит аминокислота j).
Как это работает?
Первая итерация идентична BLASTP. Затем генерируется множественное выравнивание
последовательностей с самым высоким e-value (порог можно задать самостоятельно) и рассчитывается
позиционная матрица. Она вылавливает консервативный паттерн, который записывается как
новая матрица, в ней каждой позиции присвается оценка её консервативности.
Затем производится повторный сеанс поиска, только последовательности отбираются
по тому, соответсвуют ли они полученному профайлу. Затем из них отбираются последовательности
с максимальным e-value, и всё повторяется заново. И так до момента, когда ни одна найденная новая последовательность не превзойдёт порога.
Главный вопрос: зачем это всё?
В эволюции 3D структура белков может быть консервативна даже при условии
значительного расхождения последовательностей. PSI-BLAST как раз
замечает такие нюансы.
4) PHI-BLAST (Pattern Hit Initiated BLAST)
Это видоизменение PSI-BLAST, которое на вход получает определённый мотив,
вокруг которого и выстраивает позиционную матрицу.
5) DELTA-BLAST (Domain Enhanced Lookup Time Accelerated BLAST)
использует Conserved Domain Database (CDD), ресурс NCBI, позволяющий
идентифицировать консервативные домены в белковой последовательности.
Кажому домену там сопоставлено множественное выравнивание и посчитана позиционная
матрица. Благодаря использованию этой базы данных уходит потребность
первичного запуска BLASTP, увеличивается скорость и DELTA-BLAST заявлен как алгоритм, который находит
гомологов лучше, чем остальные алгоритмы.
Основные параметры:
1) max target sequences: порог количества последовательнотей, после
которого алгоритм останавливается
2) short queries: автоматически устанавливать word size
и другие параметры для небольших последовательностей
3) expect threshold: верхний порог e-value.
E-value - величина, характеризующая математическое ожидание того,
что в случайно в базе данных среди последовательностей такой же длины и
такого же аминокислотно состава найдется последовательность, дающая такой же или более вес.
Соответсвенно, чем меньше e-value, тем больше мы можем доверять выравниванию.
4) word size:
длина k-меров, на которые разбиваются последовательности из баз данных и данная последовательность.
В случае нуклеотид-нуклеотидного бласта для начала построения выравнивания требуется
полное совпадения k-меров, в других разновидностях бласта неточные совпадения рассматриваются
с точки зрения схожести двух последних, в результате делается вывод, выравнивать или нет.
5) max matches in a query range:
эта опция полезна, если множество сильных совпадений с одной частью последовательности
может помешать BLAST найти более слабые совпадения с другой частью последовательности.
Параметры оценки:
1) matrix:
на выбор даются матрицы BLOSUM(INT) и PAM(INT). В случае BLOSUM число означает, что
матрица была посчитана для последовательностей с меньшей, чем (INT), similarity. В случае PAM -
PAM(INT) - это матрица, соответсвующая временному интервалу,
достаточному для возникновения (INT) мутаций на 100 аминокислот.
2) gap costs: различия между различными видами штрафов знакомы из прошлого практикума.
Надо помнить, что повышение gap opening penalty повышает частоту инделов в выравнивании,
а gap extension penalty влияет на их размеры.
3) compositional adjustments:
матрицу можно дополнительно улучшить. Сomposition-based statistics - самый простой подход,
нормирует все scores для мест замен определённой аналитически константой, оставляя
все scores с гэпами фиксированными. Этот подход универсален, а следующий,
более замысловатый - compositional score matrix adjustment - выгодно применять
только в определённых случаях. Он настраивает
каждую оценку в матрице подстановок отдельно,
чтобы компенсировать композиции двух сравниваемых последовательностей.
Фильтры и маски:
1) filter:
low-compleqity фильтр нужен для того, чтобы биологический смысл полученных результатов
превалировал над статистическим. Данная опция скрывает участки с высокой степенью вырожденности,
например, гомополимерные участки, короткие тандемные повторы и перепредставленность определённых
аминокислотных остатков.
2) mask:
mask for lookup table only: применяется на этапе составления таблицы поиска
для того, чтобы бласт не выдавал результатов с теми же участками с высокой степенью вырожденности.
mask lower case letters: маскирует все аминокислоты, введённые в нижнем регистре.
Поиск гомологов
Я провела три сеанса поиска в BLAST, во всех трёх я
изменила базу данных на swissprot и ограничение на число находок на 20000.
Мне было интересно, будут ли значительно различаться выдачи разных алгоритмов, поэтому
я провела сеансы поиска с blastp (по умолчанию), PSI-BLAST и DELTA-BLAST.
В случае DELTA-BLAST средний показатель identity был самым низким (в районе 30%, в то время как для других алгоритмов - 37%).
Однако alignment scores получились в среднем сильно больше - порядка 500 (причём scores первого и, например, десятого
объекта выдачи различаются на 17 баллов), в то время как для двух других
показатели score очень похожи: первый результат выдачи имеет score 766 и отличается от
десятого на 495 баллов.
Скорее всего такое сильное расхождение результатов объясняется моими малыми познаниями,
так как получение адекватных результатов из DEPLTA-BLAST
"требует значительных знаний и хорошей интуиции, а поэтому запуск таких иттеративных
алгоритмов - более сложная задача, чем просто запустить blastp" (ИР, большое спасибо за ответ).
Поэтому дальнейшая работа производилась с результатами blastp.
Таблицы
BLASTp
PSI-BLAST
DELTA-BLAST
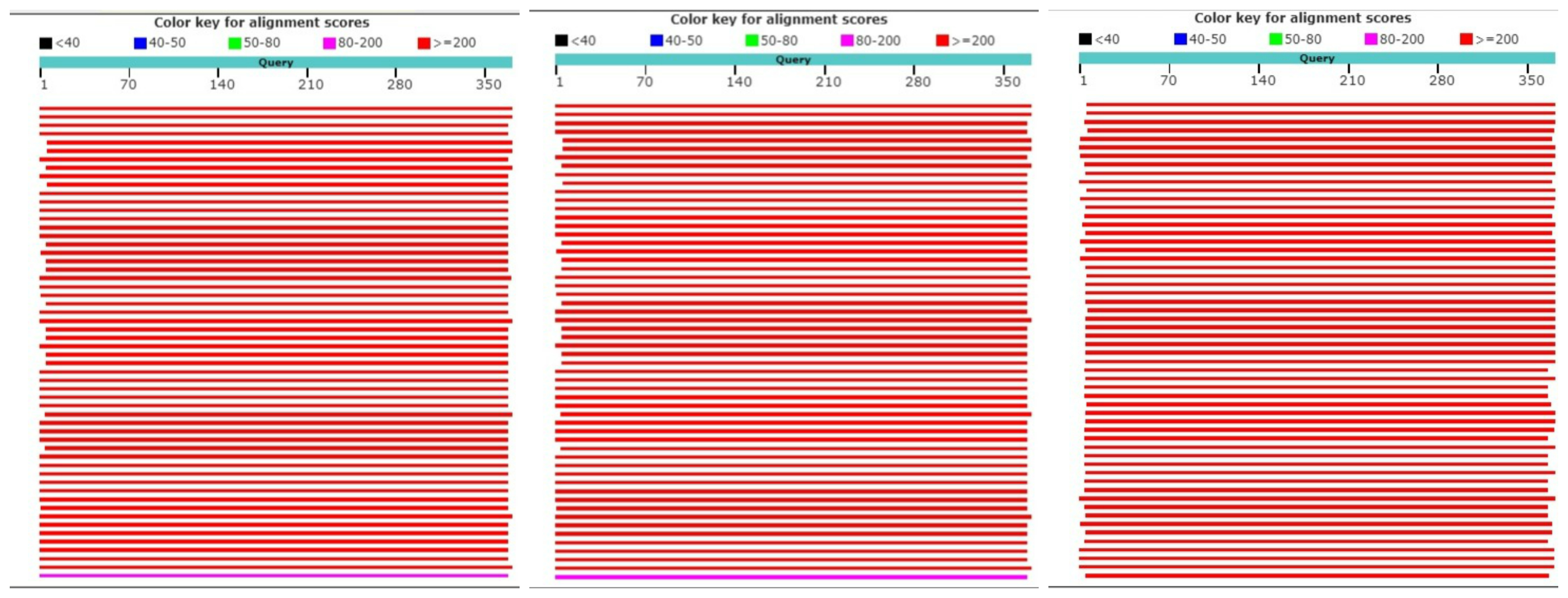 Рис.1 Результаты работы BLAST, слева направо: blastp, PSI-BLAST, DELTA-BLAST
Рис.1 Результаты работы BLAST, слева направо: blastp, PSI-BLAST, DELTA-BLAST
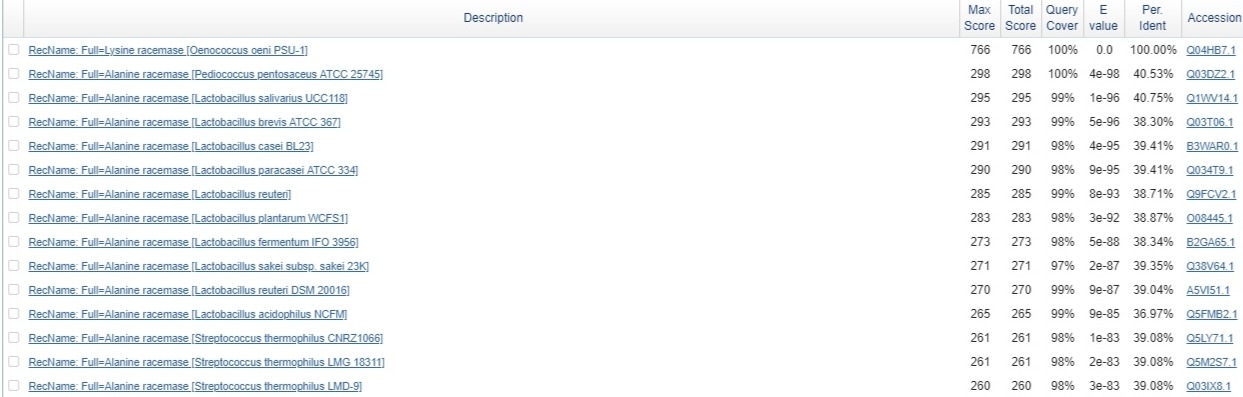
Рис.2 Выдача BLASTp
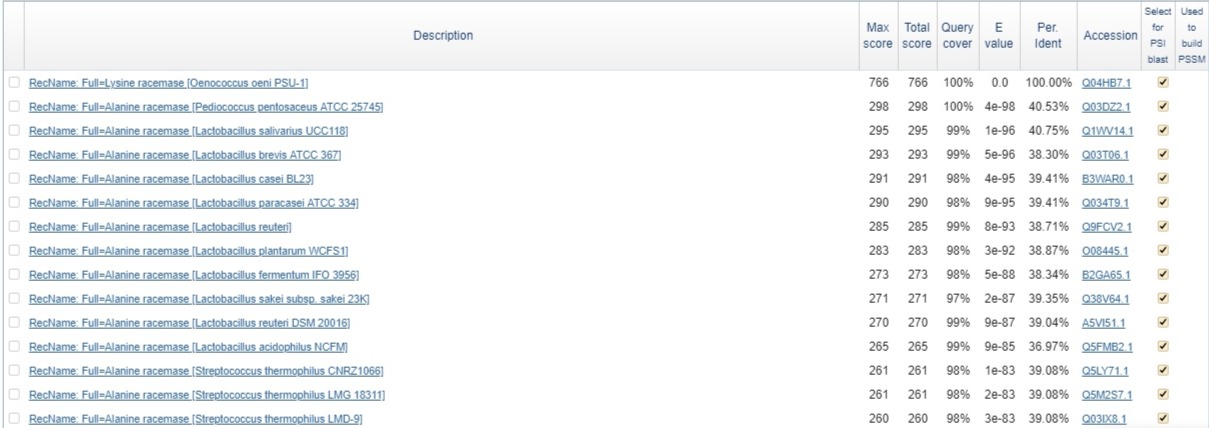
Рис.3 Выдача PSI-BLAST
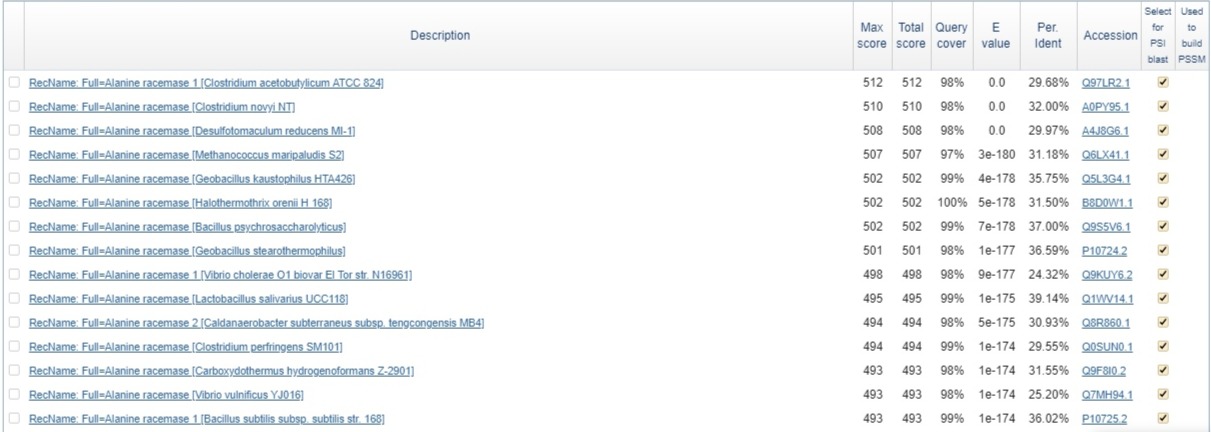
Рис.4 Выдача DELTA-BLAST
Первый результат выдачи с E-value 0 - сам белок.
Забавно, что единственная лизин-рацемаза в выдаче - как раз данный белок Oenococcus oeni.
Все наиболее близкие к нему белки носят название аланин-рацемаз. Данный вопрос
затрагивался мной при выполнении практикумов в прошлом семестре, интересно, что
аланин-рацемаза может катализировать
рацемизацию и аланина, и лизина, а лизин-рацемаза - только лизина.
Посмотрим же на выравнивания, чтобы понять, почему так.
Выравнивания
Одна из последовательностей была взята из Geobacillus stearothermophilus, потому что
аланин рацемаза данного организма сравнивается с рассматриваемой
лизин-рацемазой в статье [2], которую я разбирала для практикума в прошлом семестре.
В таблице 1 представлены все последовательности, бравшиеся для выравнивания.
Указанные в статье изменённые остатки в активном центре действительно
отличаются для лизин-рацемазы, причём у всех остальных найденных белков подобных замен нет.
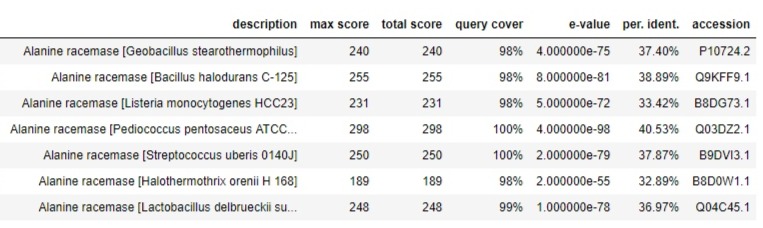
Таблица 1. Последовательности, бравшиеся для выравнивания.

Рис. 5. Выравнивание. Простите, с css я раздружилась за долгое время, а
всё это красивое выравнивание хочется показать целиком.
Скачать jalview проект
Данные белки гомологичны, так как присутствует блок (рис. 6) длины 7,
начинающийся и заканчивающийся абсолютно консервативной
позицией, без колонок с гэпами, и в котором высока плотность
консервативных позиций.
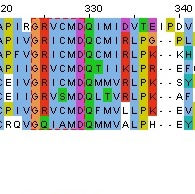
Рис. 6. Блок выравнивания.
Итак, какие выводы о лизин-рацемазе мы можем сделать на основании данного выравнивания?
Построим дерево по методу Neighbourhood joining с использованием BLOSUM62 (рис. 7).
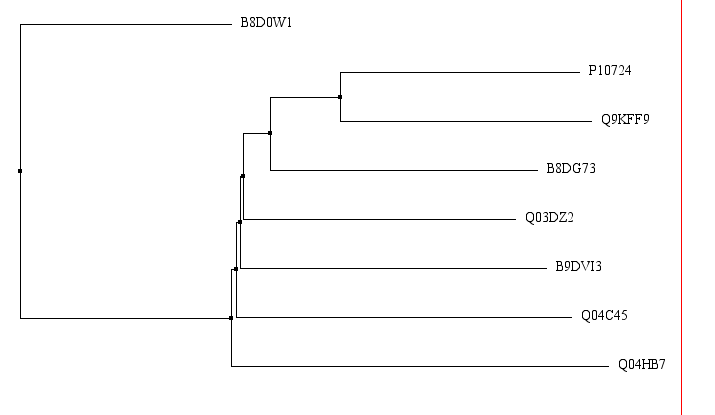
Рис. 7. Дерево, построенное на основе выравнивания.
Интересно, что изучаемая лизин-рацемаза (Q04HB7) отстоит от большинства
остальных рацемаз, следовательно,
отделение лизин-рацемазы от ветви аланин-рацемаз произошло довольно рано.
Однако самым дальним белком является аланин-рацемаза Halothermothrix orenii из класса Clostridia,
что закономерно, так как все остальные белки принадлежат бактериям из класса Bacilli.
Карта локального сходства белков
Были рассмотрены белки с идентификаторами A0A163IMY9_DIDRA и FOL1_DICDI. Первый -
дигидронеоптерин-альдолаза (dihydroneopterin aldolase) аскомицета Didymella rabiei,
участвующая в процессе синтеза фолата. Второй белок - FOL1 (фолат-синтезирующий белок)
Dictyostelium discoideum
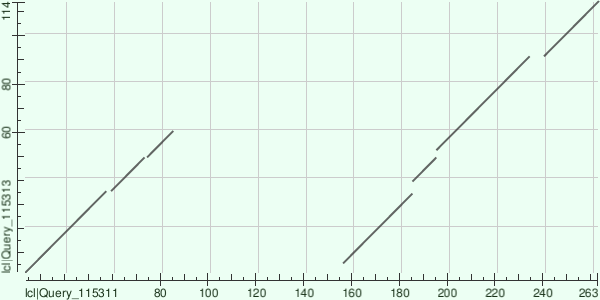
По вертикали расположена дигидронеоптерин-альдолаза (268 а.о.), по горизонтали -
FOL1 (657 а.о.). Видна дупликация в FOL1 и мелкие инделы. Выравниваются начало
дигидронеоптерин-альдолазы и середина FOL1. e-value для двух полученных выравниваний
довольно мал, поэтому оба выравнивания использовались для анализа.
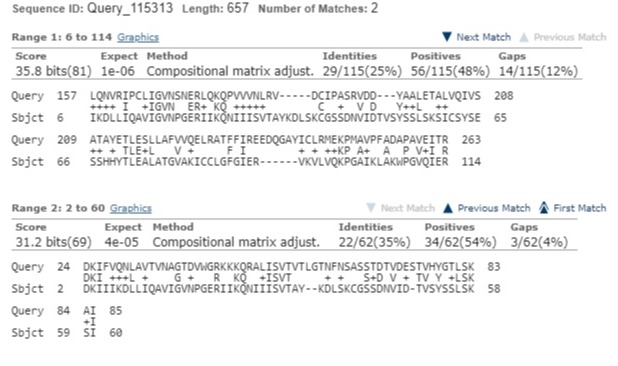
Игры с BLAST
Последовательность, по которой производился поиск: GIVEPEACEACHANCE.
На рис. 6 видно, что эта последовательность символов не является естественной для живых организмов.
Только АТФ-зависимая рРНК хеликаза RRP3 из паразитического
гриба Pyricularia oryzae показала 83.33% identity, хотя все остальные
показатели плохие, и эти белки нельзя считать гомологичными.

Рис. 6. Результаты для последовательности GIVEPEACEACHANCE.
Затем я дала на вход монолог Лаки из "Ожидания Годо"[3] без пробелов и с
символами высокого регистра
(2751 символов, были убраны буквы, не соответсвующие аминокислотам). Не было выдано никаких результатов,
этот монолог даже в виде набора аминокислот бессмысленен.
Однако если уменьшить параметр word size до трёх, будет найдено 4 соответствия.
Между прочим, одно из них принадлежит автотранспортеру ROD_p1121 (D2TV88).
Это белок, находящийся на мембране Citrobacter rodentium, и total score для него
составляет 78.9, а e-value 0.052. Всё же показатели max score, query cover и per. ident.
для данного белка неубедительные. Были найдены негомологичные белки
из разных групп организмов, например, один из четырёх принадлежит Homo sapiens.
[1] http://mirrors.vbi.vt.edu/mirrors/ftp.ncbi.nih.gov/blast/documents/researchfestivalpages.pdf
[2] Shiro Kato, Hisashi Hemmi, Tohru Yoshimura; Lysine racemase from a lactic acid bacterium, Oenococcus oeni: structural basis of substrate specificity, The Journal of Biochemistry, Volume 152, Issue 6, 1 December 2012, Pages 505–508, https://doi.org/10.1093/jb/mvs120
[3] https://resources.saylor.org/wwwresources/archived/site/wp-content/uploads/2011/01/Waiting-for-Godot.pdf