создание файла с полиморфизмами в bcf формате. документация samtools
угрожает перестать поддерживать данную опцию и предлагает файлы bcf генерировать
при помощи bcftools mpileup, а samtools mpileup использовать для получения
файлов pileup формата.
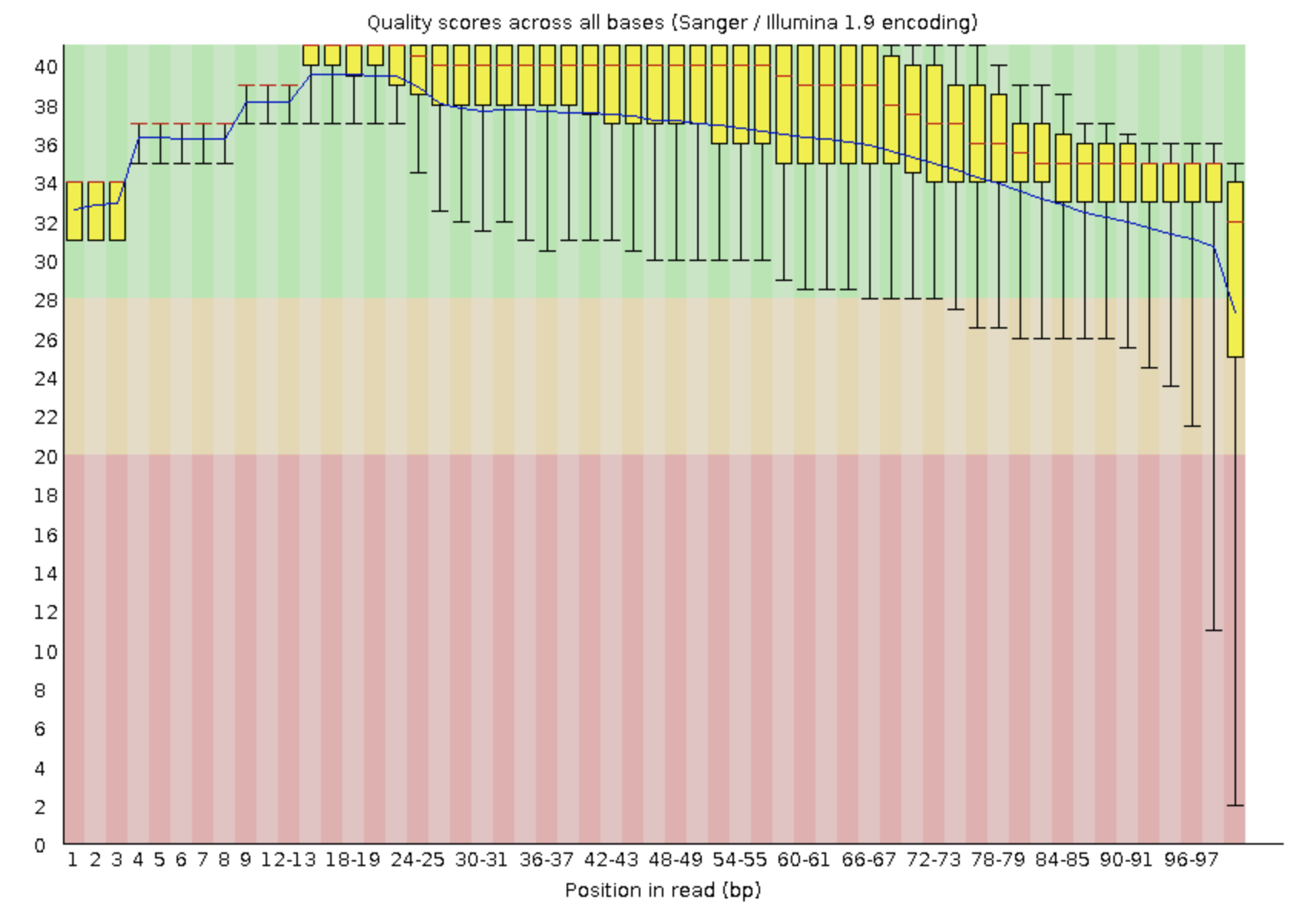
Приведите картинку из результатов FastQC с оценкой качества Ваших чтений
Рис. 1.
качество прочтений до тримминга
Рис. 2.
качество прочтений до тримминга
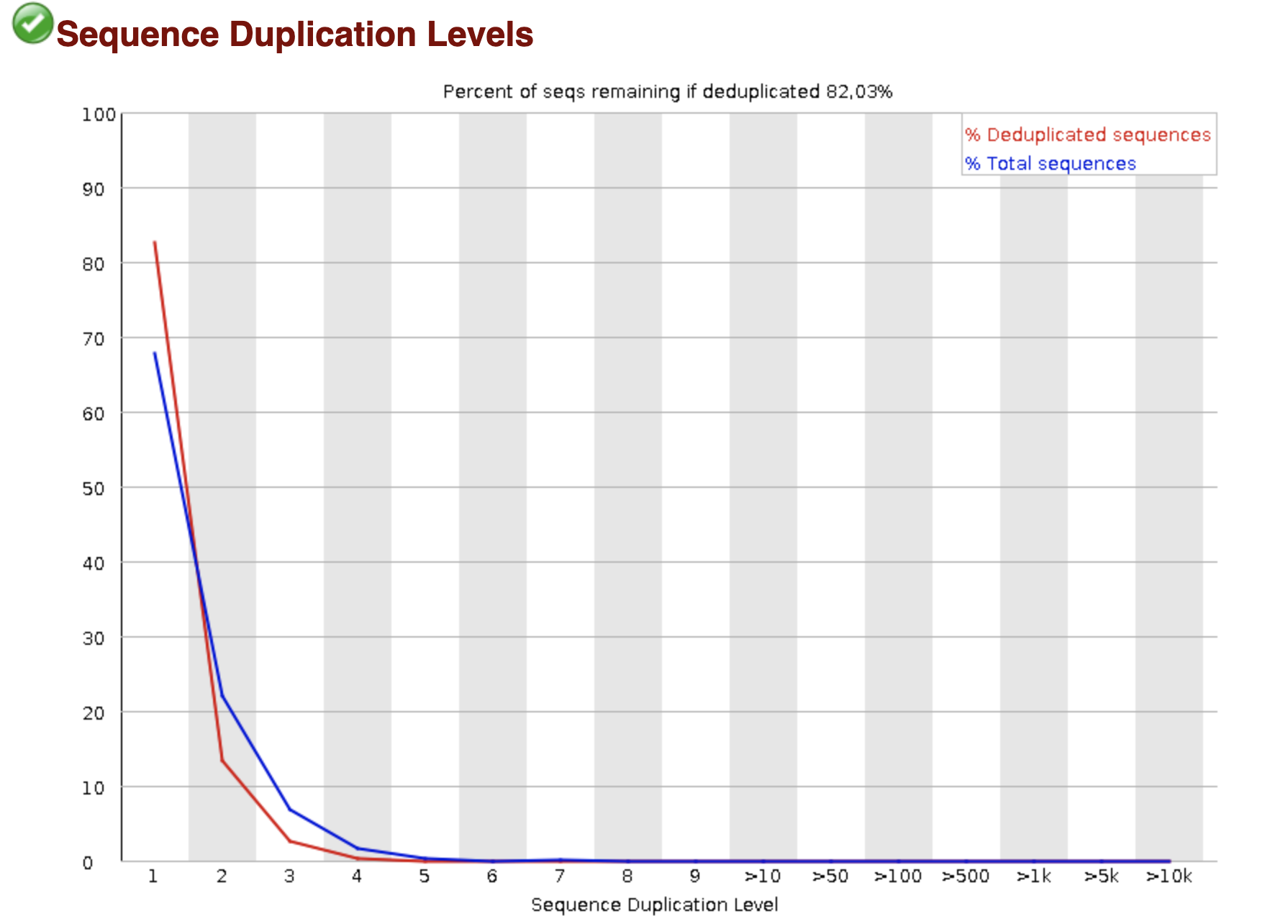
Доп: Приведите и объясните еще любую картинку из результатов FastQC, используя
пояснения в руководстве к программе.
Рис. 1.
уровень дупликации последовательностей
Дупликаты на уровне сырых прочтений - идентичные последовательности.
Красная линия: распределение уровней дедупликации полного набора
последовательностей.
Синяя линия: последовательности дедуплицируются, то есть по питоновской аналогии
список превращается в множество, а затем считается отношение количества
последовательностей из дедуплицированного набора к количеству
последовательностей до дедупликации. Надеюсь, я правильно поняла.
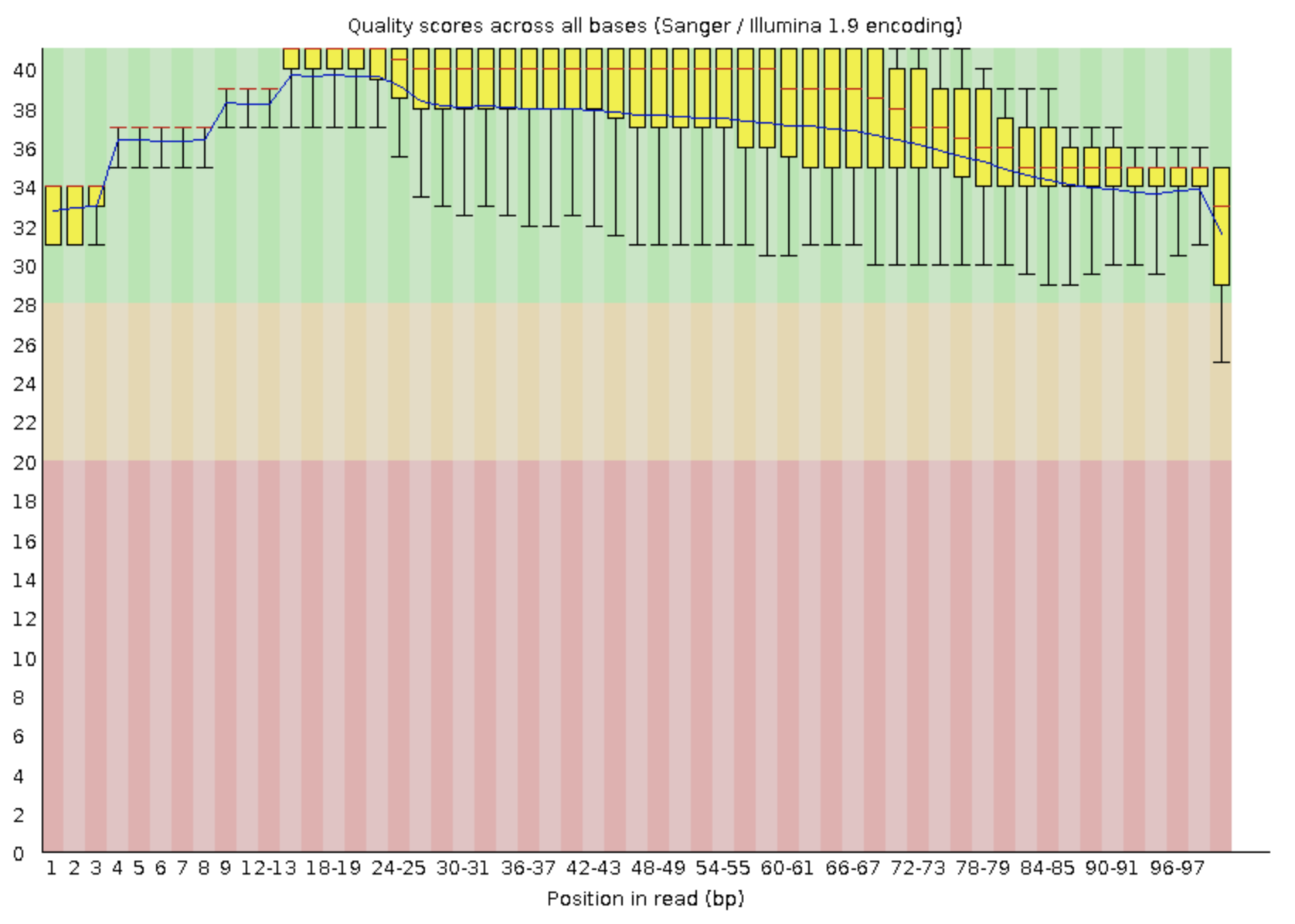
Сколько чтений осталось после процедуры триммирования? Оправдано ли
триммирование в случае Ваших чтений или можно было обойтись без этого?
Аргументируйте. Приведите картинку качества чтений(FastQC) после триммирования.
После триммирования из 8696 прочтений выжило 8562 (98,46%), было отброшено 134
(1,54%). Если посмотреть на анализ качества до и после триммирования, можно
увидеть, что отрезались как раз нуклеотиды с плохим качеством, которые могли бы
нам помешать корректно картировать риды.
Сколько чтений (%) картировано на геном?
99.75%
Сделайте вывод о качестве картирования.
Картирование очень хорошее, так как с такими строгими параметрами только 21
чтение из 8562 не картировалось ни разу и только 2 чтения были картированы более
одного раза.
Описание трех полиморфизмов из .vcf файла
Хромосома 14
Позиция 81448951: GAAAAAAAAAA => GAAAAAAAAAAAA,GAAAAAAAAAAA,GAAAA
AAAAAAAAA (инсерция), глубина=55, качество=84.4632
Позиция 81467864: CAT => C (делеция), глубина=16, качество=217.468
Позиция 81448282: C => G (замена), глубина=5, качество=91.0145
Сколько snp и сколько инделей Вы получили?
87 SNPs (63 транзиций и 24 трансверсии) и 5 инделей
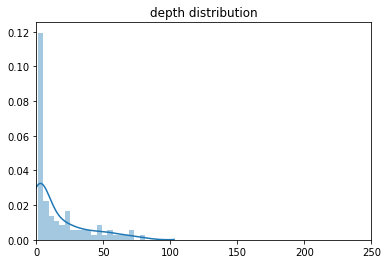
Хорошее ли покрытие и качество у найденных полиморфизмов (приведите
распределение)?
Глубина в основном довольно низкая. Качество варьирует.
На какие категории делит snp база данных refseq в annovar? Сколько snp у Вас
попало в каждую группу?
exonic(3)/intronic(85)/splicing(1)/UTR3(1)/intergenic(0)/ncRNA(0)/UTR5(0)/upstream(0)/downstream(0)
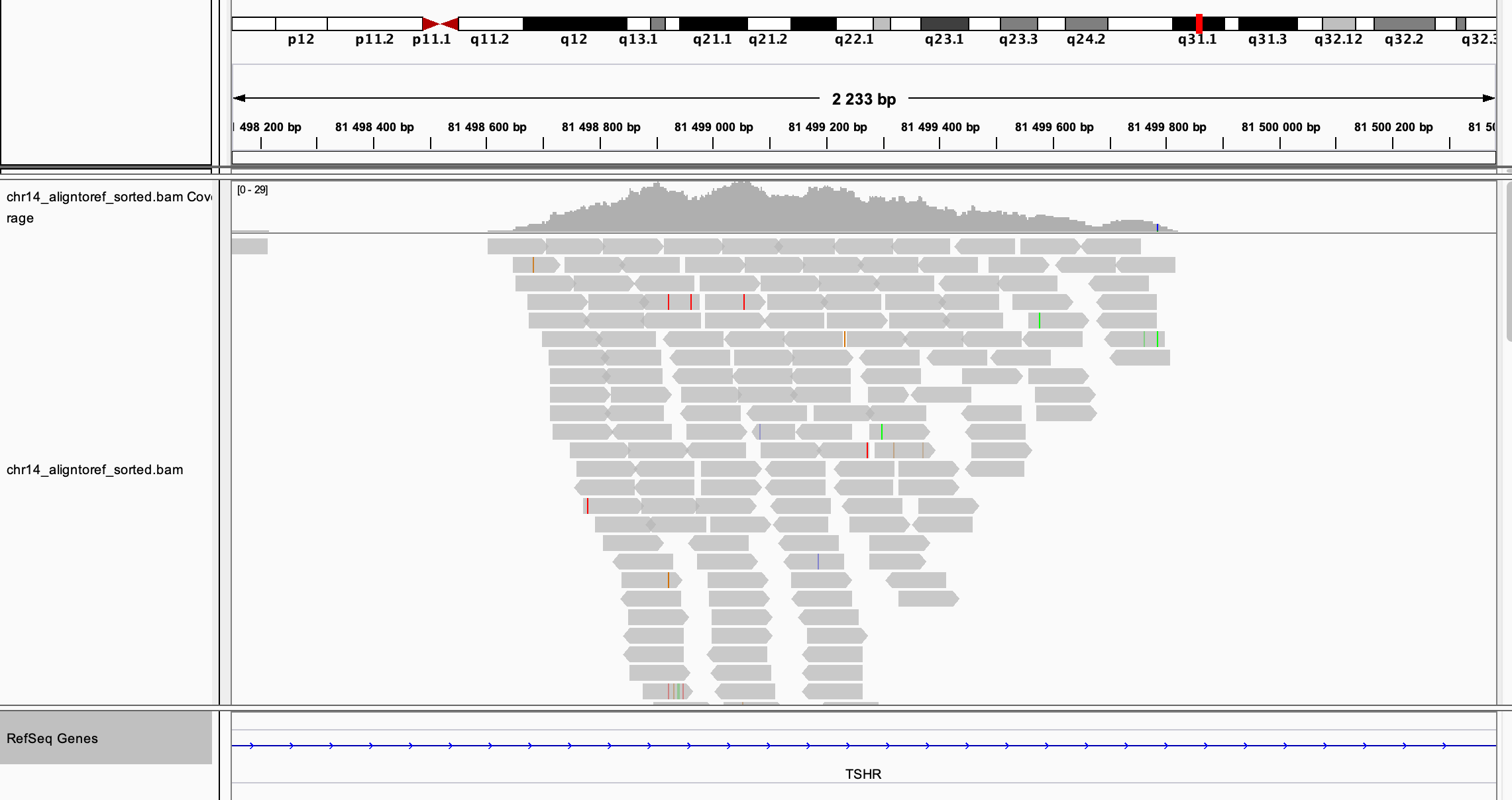
В какие гены попали Ваши snp?
RNASE9, TSHR, PPP2R5C. Иллюстрация для TSHR из IGV:
К каким нуклеотидным и аминокислотным заменам привели snp?
RNASE9: A => G (T => C), TSHR: T => C (T => C), PPP2R5C: G => C (A => P).
Сколько snp имеет rs?
83 из 90, согласно dbsnp
Что Вы можете сказать о частоте найденных snp?
0.255471 - средняя частота, рассчитаная по выдаче 1000genomes. Если minor allele
frequency (MAF) < 0.01%, то аллель считается редким. Как мы видим, в основном
полученная выборка состоит из довольно частых SNP.
Что Вы можете сказать о клинической аннотации snp?
По аннотации gwas, три SNP имеют клиническое значение. Аутизм (CINP-участник
рекликации ДНК, регулирует сигнализацию о прохождении контрольных точек
клеточного цикла), рак простаты и
(NDRG2-регулятор Wnt пути, в норме tumor suppressor), Базедова болезнь.
clinvar ничего не нашёл