индексирование хромосомы, не допускаются частичные
несовпадения (--no-softclip), параметр --no-spliced-alignment для рнк-сека не
используется, так как имеем дело со зрелыми мРНК
htseq-count -f bam chr14_rna1_aligntoref_sorted.bam -s no
/nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf
Запуск htseq-count с параментрами: -f bam, то есть на вход подаётся
bam-файл, -s no, специфично ли было секвенирование по направлению, -i и -m по умолчанию,
потому что gene_id - это удобно, и метод - union, чтобы допускать выход рида за
пределы гена и не допускать однозначной аннотации рида, лежащего на
перекрывающихся генах.
cat chr14_rna1_count.txt | awk '$2 != 0'
подсчёт генов с ненулевым покрытием
Сколько исходно чтений Вы получили? 18189 и 17363
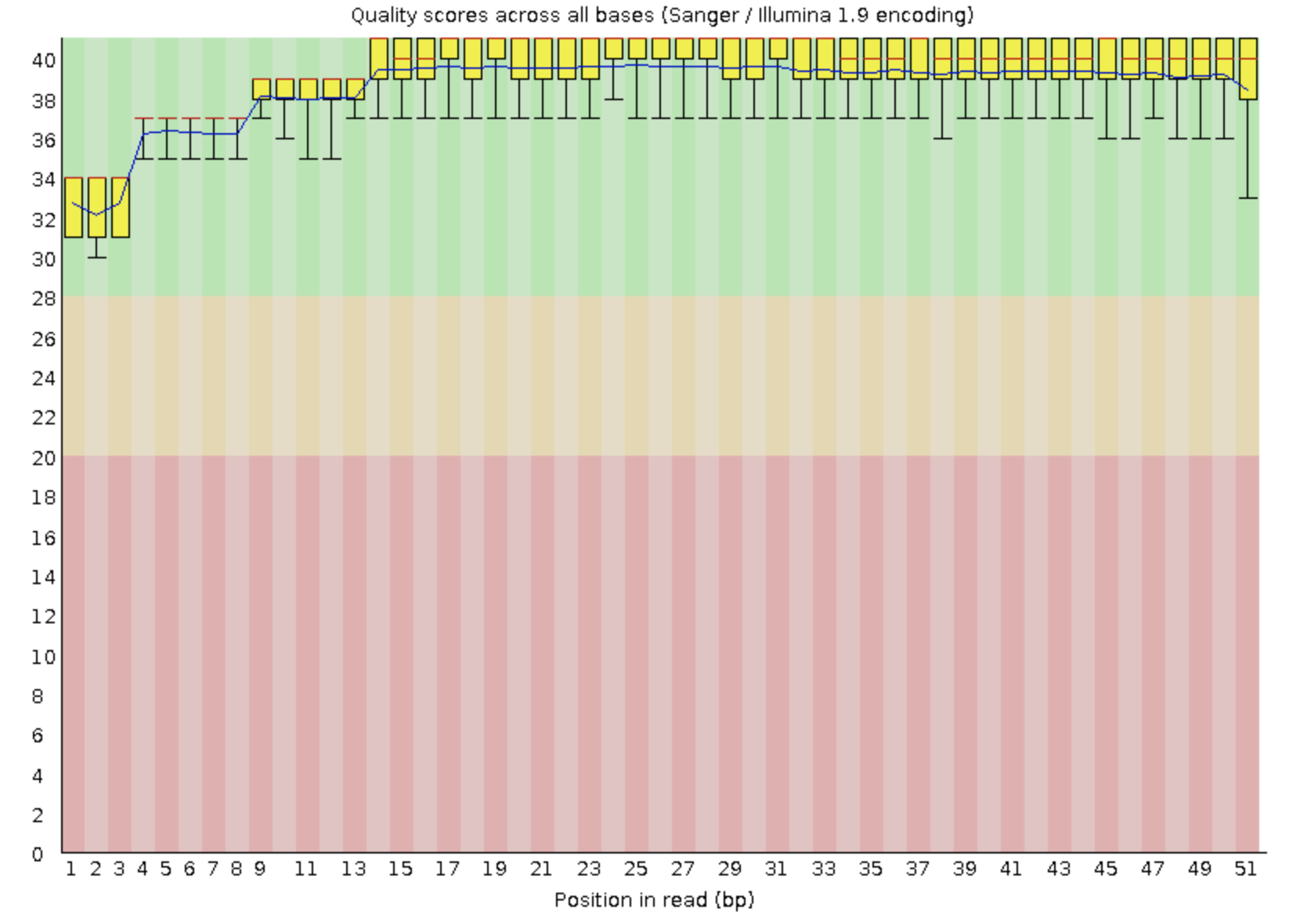
Приведите картинку из результатов FastQC с оценкой качества Ваших чтений
Рис. 1.
качество прочтений первой реплики
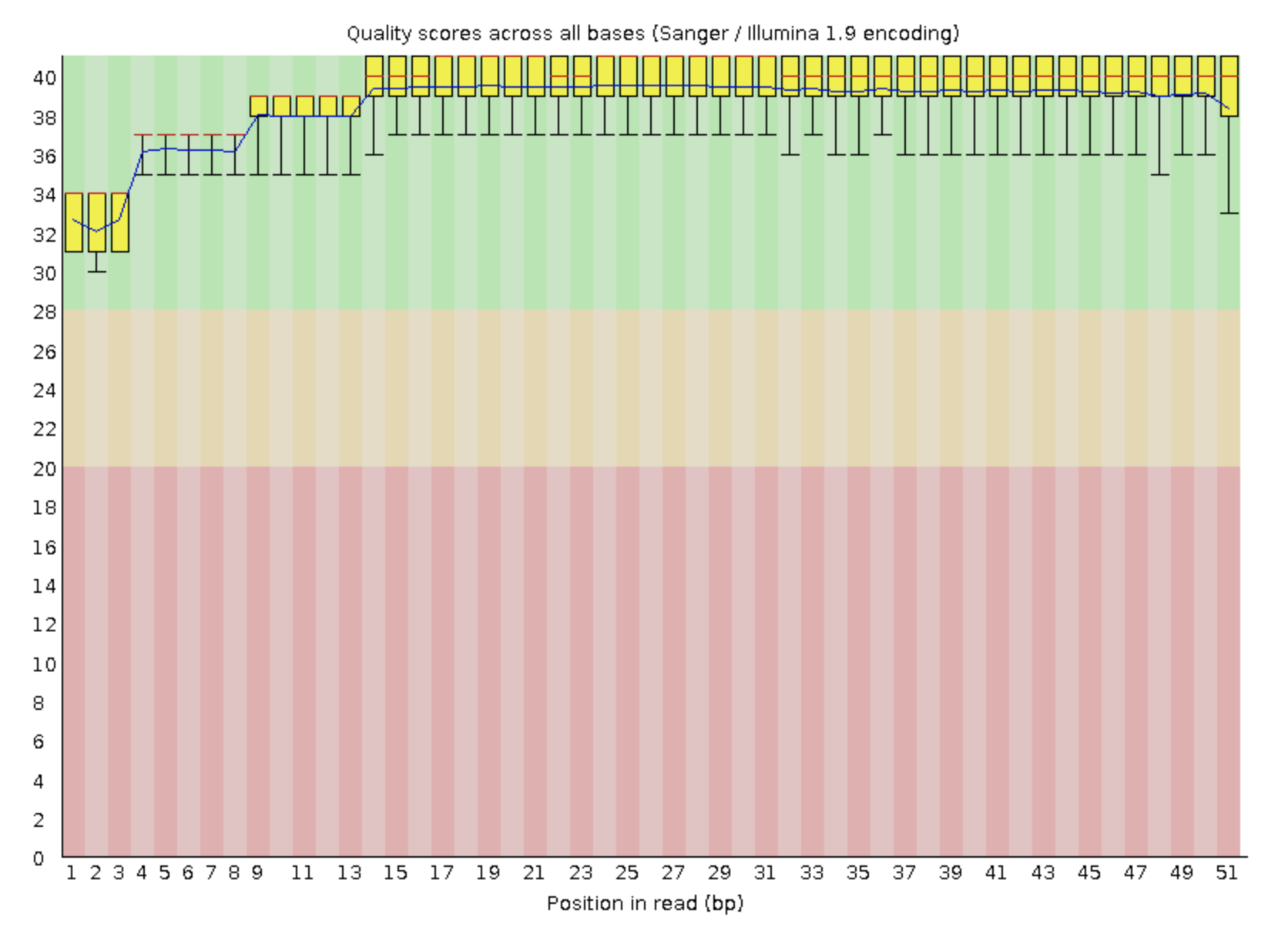
Рис. 2.
качество прочтений второй реплики
Оправдано ли
триммирование в случае Ваших чтений или можно было обойтись без этого?
Триммирование не проводилось, так как у обеих реплик качество довольно хорошее.
Сколько чтений (%) картировано на геном?
99.74%
Сделайте вывод о качестве картирования.
Картирование очень хорошее, так как только 47 (43 для второй реплики)
чтение из 18189 (17363) не картировалось ни разу и ни одно чтение (в обоеих
репликах) не было картировано более
одного раза.
Анализ результатов
ген ENSG00000080824.14 - ген белка HSP90AA1, белок теплового шока 90, в первом случае
он составляет 18115 прочтений (из 18189, 99.5%), во втором - 17287 (из 17363, 99.6%).
Мы не знаем, как проводилось секвенирование, но скорее всего, это был не тотальный рнк-сек :) Поэтому сравнивать
лучше не процентные соотношения, а общее количество прочтений в двух репликах. Видимо, в первой реплике эксперимента
температура была выше.
Прочтения, которые "не нашли своего места" и никуда не маппировались,
возможно, слишком короткие. Собственно, если вернуться к результатам fastqc,
видно, что с адаптерами всё в порядке (их совсем нет), но зато
k-mer content довольно плох. Есть короткие часто встречающиеся последовательности.
В феврале 2019 года вышел фреймворк Galaxy, CAFU, в статье о котором
заявляется, что с такой проблемой не маппированных коротких ридов он умеет
справляться. Правда, не знаю, насколько хорошо.
Проверка других методов
Сначала подумаем, что мы хотим увидеть.
После применения intersection_nonempty мы хотим увидеть увеличение числа
картированных ридов, если ген нашего белка теплового шока пересекается с каким-нибудь
другим геном.
Далее после применения intersection_strict нам надо ориентироваться на два
предыдущих результата. Выход чтений за пределы гена уменьшит число
картированных ридов, но этот эффект может нейтрализоваться за счёт того,
что мы разрешаем нашему гену пересекаться с каким-либо другим.
Смотрим.