Находим SMILES нотацию для NAG. Это удобно сделать на странице структуры 1lmp. Сохраним эту нотацию в файл nag.smi.
C помощью obgen построим 3D структуру этого сахара в pdb формате.
obgen nag.smi > nag.mol babel -imol nag.mol -opdb nag.pdb
Скриптом prepare_ligand4.py из пакета Autodock tools создаём pdbqt файл лиганда.
prepare_ligand4.py -l nag.pdb
Так же, скриптом prepare_receptor4.py из пакета Autodock tools создаём pdbqt файл белка.
prepare_receptor4.py -r prot.pdb
Итак у нас есть входные файлы. Теперь надо создать файл с параметрами докинга vina.cfg. Для докинга необходимо указать область структуры белка в которой будет происходить поиск места для связывания. Удобно его задать как куб с неким центором. Координаты центра мы определим из модели комплекса, которую мы построили на прошлом занятии. Выберим атом сахара С2, так как он находится в центре сайта связывания и из текста pdb файла извлечём его координаты.
Построим файл vina.cfg.
Теперь можно провести первый докинг:
vina --config vina.cfg --receptor prot.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.log
Просмотрим файл nag_prot.log. Энергии 3ёх лучших расположений и геометрическую разницу между ними:
mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+---------- 1 -5.8 0.000 0.000 2 -5.5 2.763 4.413 3 -5.4 1.759 2.952В PyMol загрузим файлы nag_prot.pdbqt и prot.pdbqt. Отобразим все состояния на одной картинке.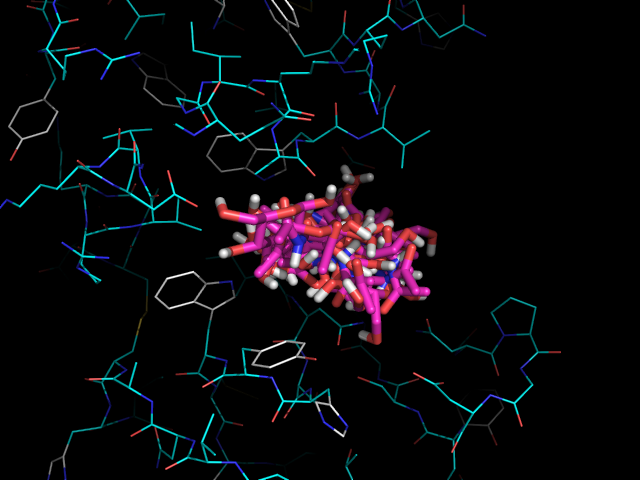
Здесь мы видим, что лиганду доступен некоторый объём в центре связывания и он, возможно, связывается с белком не очень жёстко.
Теперь проведём докинг рассматривая подвижность некоторых боковых радикалов белка. Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты которые вы использовали в прошлом задании для позиционирования лиганда.
prepare_flexreceptor4.py -r prot.pdbqt -s ASN64_TRP82_ASP120
и проведём докинг:vina --config vina.cfg --receptor prot_rigid.pdbqt --flex prot_flex.pdbqt --ligand nag.pdbqt --out nag_prot_flex.pdbqt --log nag_prot_flex.log
Время счёта было в несколько раз больше, чем для случая с жёстким белком. Просмотрим файл nag_prot_flex.log. Энергии 3ёх лучших расположений и геометрическую разницу между ними:
mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+---------- 1 -5.1 0.000 0.000 2 -4.8 0.956 1.991 3 -4.7 1.620 3.076
По сравнению с первым случаем, у лиганда здесь гораздо больше возможностей связаться с белком. Здесь лиганд в различных конформациях занимает больший объём. Слева снизу на картинке даже есть одна обособленная конформация лиганда. Но смущает тот факт, что энергия лучшего связывания с некоторыми подвижными аминокислотами (-5.1) гораздо выше энергии связывания с жёстким белком (-5.8).
Расположение, наиболее близкое к тому, что было получено в моделировании, находится на 18 месте в списке для конформаций с жёстким белком, с энергией связываения -4.4 ккал/моль. Но в данном случае и не следовало ожидать близких расположений, так как в при моделирование в качестве лиганда мы имели трисахарид, а при докинге всего лишь моносахарид.
NAG содержит в себе СH3C(=O)NH группу. Создадим 3 лиганда где метильный радикал этой группы будет заменён на OH (nag2.smi), NH2 (nag3.smi), H (nag4.smi). Для каждого из этих лигандов проведём обыкновенный докинг. Для этого напишем следующий скрипт:
#проведение докинга for i in {2..4};do obgen nag${i}.smi > nag${i}.mol babel -imol nag${i}.mol -opdb nag${i}.pdb prepare_ligand4.py -l nag${i}.pdb vina --config vina.cfg --receptor prot.pdbqt --ligand nag${i}.pdbqt --out nag${i}_prot.pdbqt --log nag${i}_prot.log done #получение таблиц трёх лучших расположений #для каждого проведённого докинга for i in *.log;do echo -e "$i" >> logs.txt grep 'mode' $i -A 5 >> logs.txt echo -e "\n" >> logs.txt doneТаблицы лучших расположений.Для получения изображений откроем файл prot.pdbqt, расположим структуру так, чтоб по центру экрана был центр связывания, и выполним следующий скрипт в PyMOL:
load nag_prot.pdbqt, n1 load nag2_prot.pdbqt, n2 load nag3_prot.pdbqt, n3 load nag4_prot.pdbqt, n4 set all_states, on disable all enable prot.pdbqt python for i in range(1,5): cmd.enable("n"+str(i)) cmd.show("sticks","n"+str(i)) cmd.ray(640,480) cmd.png("nag"+str(i)) cmd.disable("n"+str(i)) python endПолученные изображения: nag2.png, nag3.png, nag4.png.
Для удобства анализа сделаем из них одно анимированное изображение: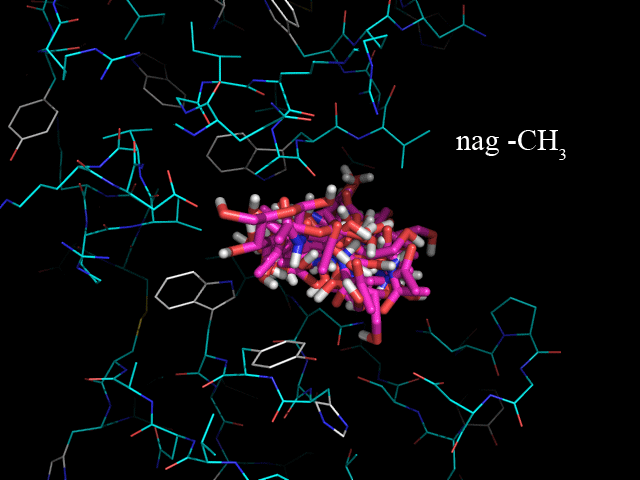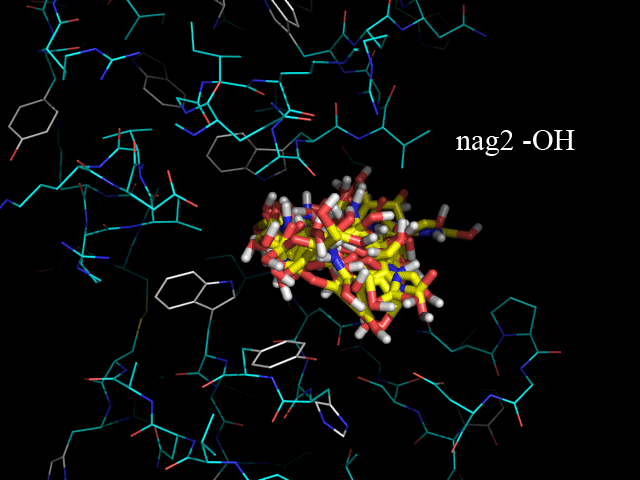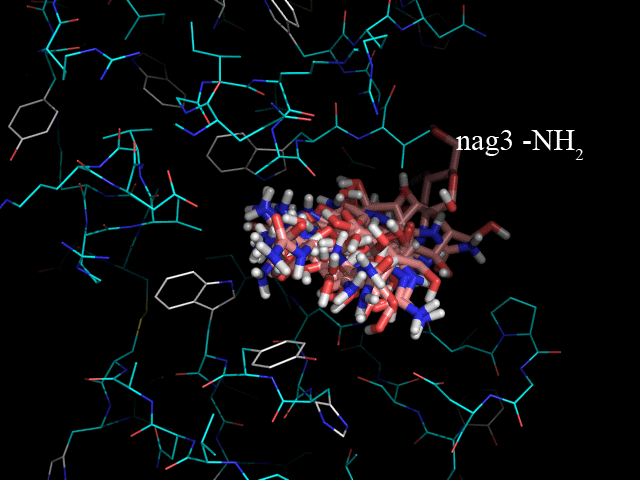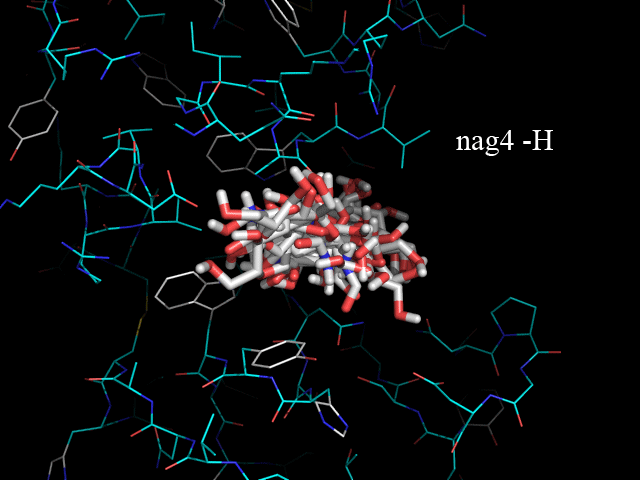
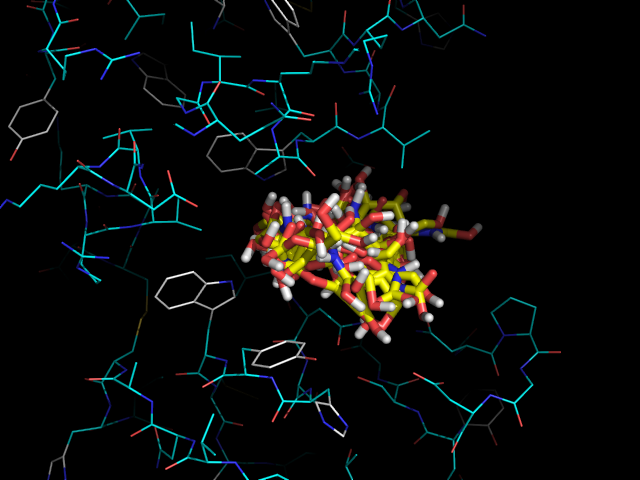
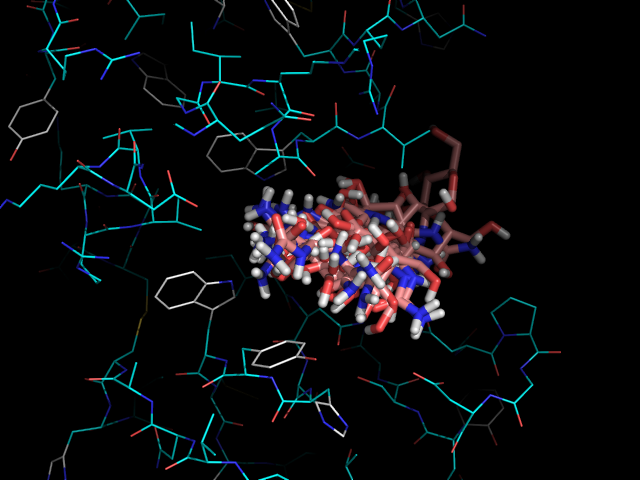
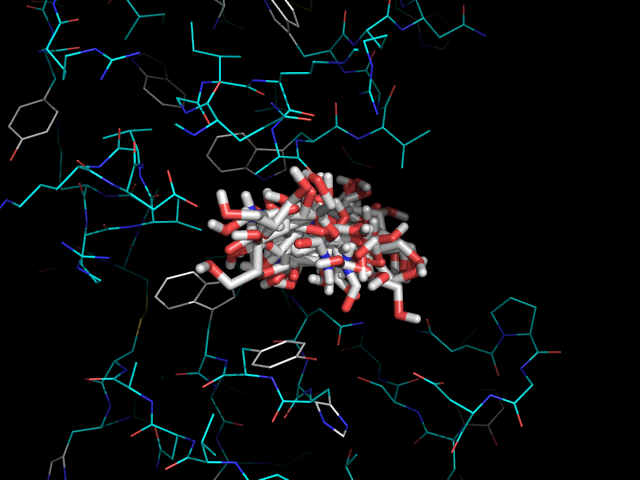
На изображении видно, что одни лиганды помещаются в белке более глубоко, а другие менее. Глубже всех связывается nag4 (-H), дальше всех - nag2 (-OH). Хотя наибольшую аффинность показывает nag3 (-NH2), -5.9 ккал/моль.
{kind=link}
{kind=link}
{kind=link}