export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Оптимизация порфирина
Cкачиваем аннотацию порфирина в виде SMILES (здесь). С помощью программы obgen и после удаления лишних водородов получаем 3D-структуру порфирина.
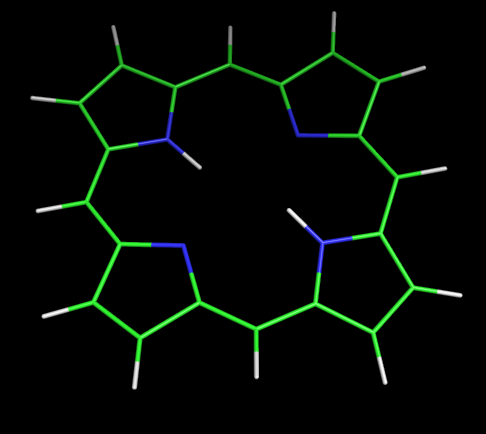
Программой babel получаем входные файлы для программы Mopac с параметризацией PM6 и AM1 и запускаем Mopac:
MOPAC2009.exe имя_файлаИ программой babel переводим выходной файл с расширением .out в PDB. Сравним полученные оптимизации.
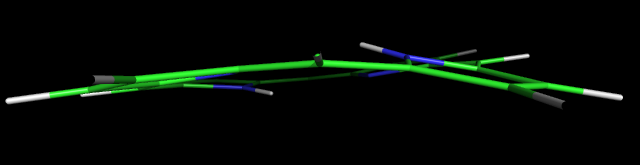


Обе параметризации дали неплохой результат, структуры выглядят плоскими. Но с AM1 Mopac справился немного лучше.
Возбужденные состояния порфирина
Для указания Mopac о необходимости расчёта возбуждённого состояния добавляем в конец файла:пустая строка cis c.i.=4 meci oldgeo some descriptionПриведена часть таблицы выходного файла с энергиями электронных переходов с добавлением столбца соответсвующих длин волн:
STATE ENERGY (EV) Q.N. SPIN Wave Length
ABSOLUTE RELATIVE (nm)
1+ 0.000000 0.000000 1+ SINGLET
2 1.914328 1.914328 1 TRIPLET 647
3 2.266757 2.266757 2 SINGLET 546
4 2.464105 2.464105 2 TRIPLET 503
5 2.825669 2.825669 3 TRIPLET 438
6 3.364329 3.364329 4 TRIPLET 368
7 3.391485 3.391485 3 SINGLET 365
8 3.669158 3.669158 4 SINGLET 337
9 3.871937 3.871937 5 SINGLET 320
The "+" symbol indicates the root used.
Оптимизация парабензохинона
Для молекулы O=C1C=CC(=O)C=C1 (парабензохинон) была определена геометрия с помощью obgen и Mopac. Затем была проведена оптимизация для дианиона этой молекулы. В первую строчку mop файла добавляем слово CHARGE=-2 и явным способом указываем (последовательность "(-)" справа от имени атома), что отрицательный заряд должен находиться на кислородах.obgen (зеленые углероды) и нейтральная молекула в Mopac (желтые углероды):
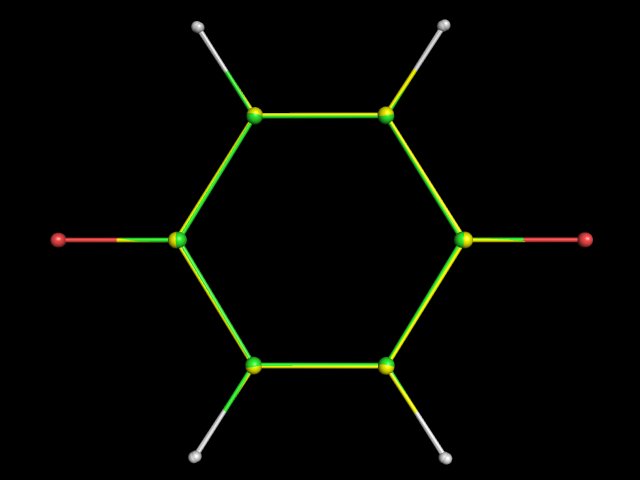
obgen (зеленые углероды) и дианион в Mopac (сиреневые углероды):
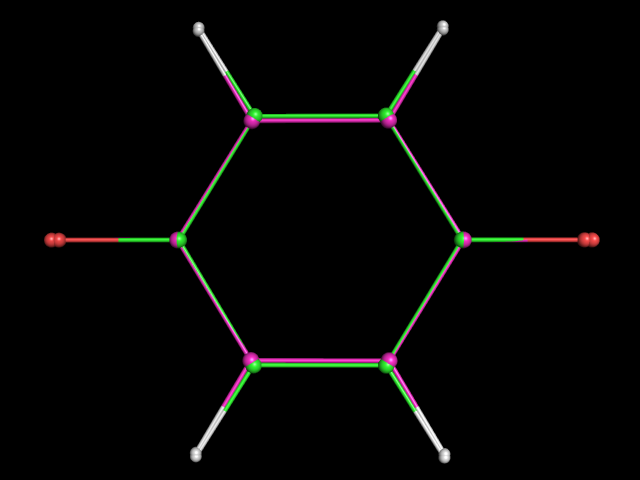
В нейтральной молекуле все атомы углерода находятся чуть дальше от центра, чем предлагает obgen. А в дианионе структура претерпевает некоторые изменения. Связи С-O удлиняются, так как из двойных становятся одинарными, а цикл становится ароматическим.
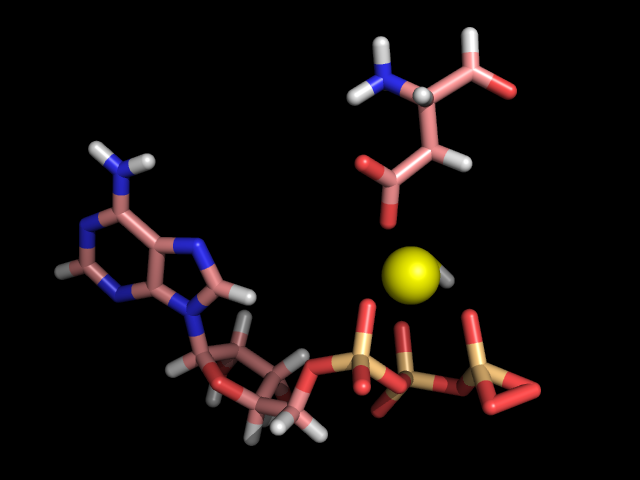
Координация магния
Дана некоторая конформация где АТФ связывается с белком через координацию иона магния, но магния в самой структуре нет. Сначала добавляем водороды с помощью babel, указывая pH среды 7.4, свойственный цитозолю клетки (опция: -p 7.4). Добавляем вручную в файл атом магния посередине между гамма-фосфором и СА аспартата. Следующим этапом указываем запрет на движение для всех атомов кроме гамма фосфата, воды и магния. Этот шаг нам нужен для того, чтобы потом легко восстановить эту конформацию в белке. Для "заморозки атомов" меняем в mop файле 1 после координаты на 0.Неоптимизированная структура
Оптимизированная структура
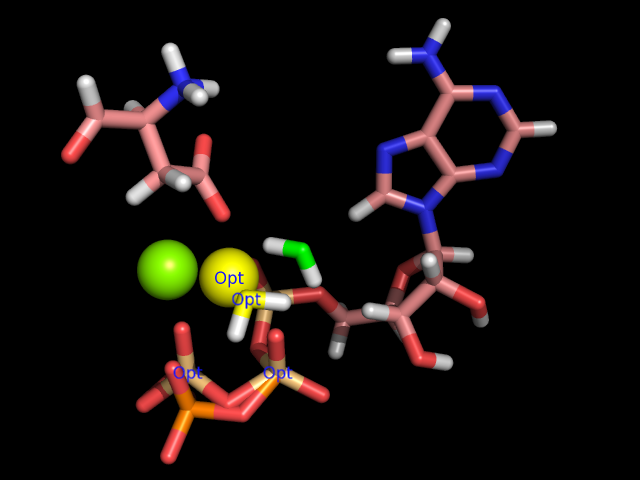
Ион магния и атомы воды покрашены разными цветами для разных структур, оптимизированные части помечены словом "Opt". Видно, что атом магния координируется кислородами трёх фосфатов, водой и кислородом аспарагиновой кислоты. Гамма-фосфат изменяет своё положение так, чтоб все три фосфата симметрично охватывали ион магния.
Сравнение с записью PDB 3PP1 (ион магния фиолетовый):
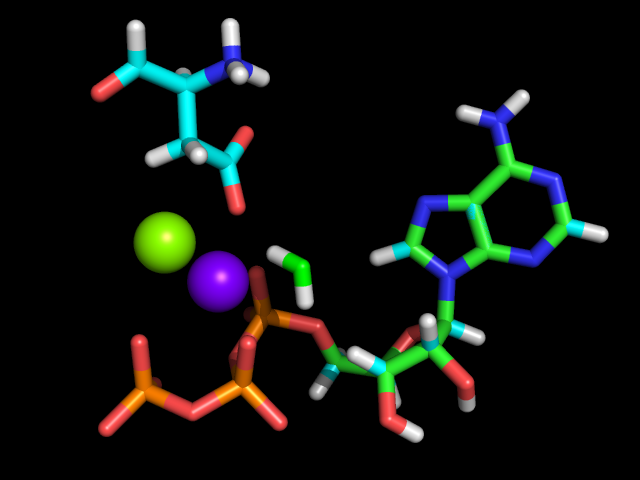
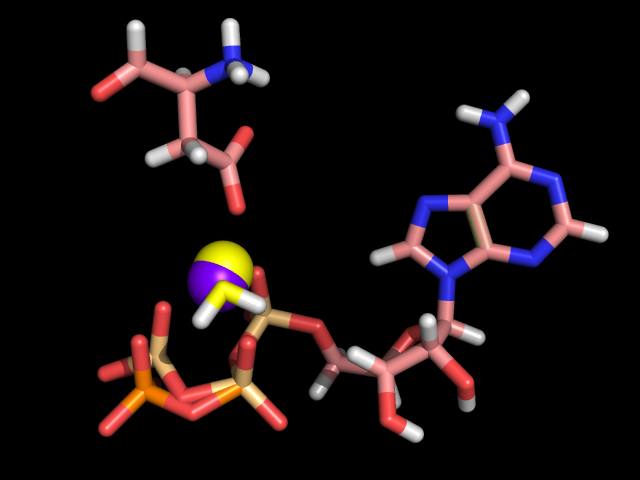
Структура молекулы АТФ в записи 3PP1 полностью совпадает со стартовой конформацией. Это довольно странно, потому что в такой конформации молекула АТФ не может координировать магний. А ион магния в этой записи имеет примерно такое же расположение, что и в полученной оптимизированной структуре.
Кроме этого была подмечена ещё одна странность. В оптимизированной структуре 2 атома кислорода на гамма-фосфате расположены настолько близко, что PyMOL рисует между ними связь. Непонятно, как такое положение может быть оптимальным.