Ресеквенирование. Поиск полиморфизмов у человека
Таблица с командами
| Программа | Что делает |
| fastqc chr13.fastq | Анализ качества чтений |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr13.fastq chr13_aftertrim.fastq TRAILING:20; java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr13_aftertrim.fastq chr13_edit.fastq MINLEN:50 | Удаляет нуклеотиды с качеством меньше 20 с конца чтений, оставляет только чтения, чья длина больше 50 |
| hisat2-build chr13.fasta chr13 | Индексирование референса (так быстрее потом картировать) |
| hisat2 -x chr13 -U chr13_edit.fastq -S chr13.sam --nosoftclip --no-spliced-alignment &> hisat2_output | Кладёт прочтения на референс (запрещаем подрезать прочтения с концов и разрывать прочтения) |
| samtools view chr13.sam -b -o chr13.bam | Преобразование полученного на предыдущем шаге sam - файла в бинарный bam - файл |
| samtools sort chr13.bam chr13_sorted | Сортировка по координате прочтения (относительно референса) |
| samtools index chr13_sorted.bam | Индексирование bam - файла |
| samtools mpileup -u -g -f chr13.fasta -o chr13.bcf chr13_sorted.bam | Поиск отличий между тем, что мы отсеквенировали, и референсным геномом |
| bcftools call -cv chr13.bcf -o chr13.vcf | Преобразование полученного на предыдущем шаге bcf - файла в vcf - файл (на него можно смотреть глазами) |
| convert2annovar.pl -format vcf4 chr13.vcf -o chr13.avinput | Конвертирование vcf-файла в файл, с которым могут работать программа annovar |
| annotate_variation.pl -out refg -build hg19 -dbtype refGene chr13.avinput /nfs/srv/databases/annovar/humandb/ | refgene - gene-based annotation |
| annotate_variation.pl -filter -out snp -build hg19 -dbtype snp138 chr13.avinput /nfs/srv/databases/annovar/humandb.old/ | dbsnp - filter-based annotation |
| annotate_variation.pl -filter -out 1000g -build hg19 -dbtype 1000g2014oct_all chr13.avinput /nfs/srv/databases/annovar/humandb.old/ | 1000 genomes - filter-based annotation |
| annotate_variation.pl -regionanno -out gwas -build hg19 -dbtype gwasCatalog chr13.avinput /nfs/srv/databases/annovar/humandb.old/ | Gwas - region-based annotation |
| annotate_variation.pl -filter -out clinvar -build hg19 -dbtype clinvar_20150629 chr13.avinput /nfs/srv/databases/annovar/humandb.old/ | Clinvar - filter-based annotation |
Часть 1: подготовка чтений
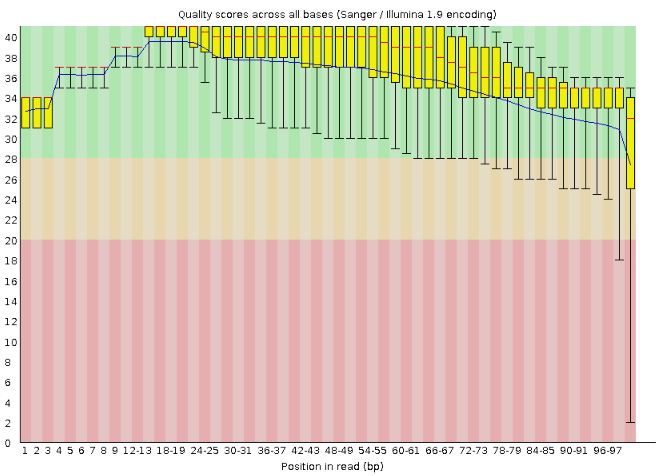
Выдача программы fastqc
До выполнения программы trimmomatic:
Количество чтений - 12155, длина - от 39 до 100.
После trimmomatic:
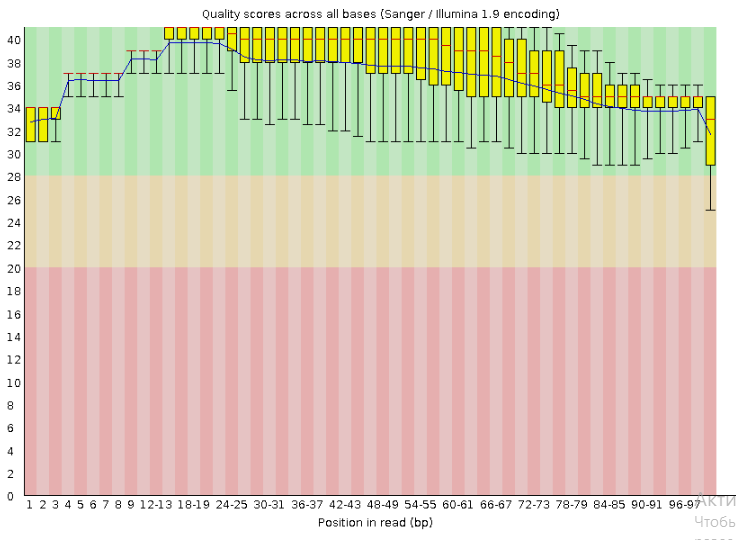
Количество чтений - 11933, длина - от 50 до 100. Видно, что качество чтений хуже всего в начале и в конце. Отрезая плохо прочтённые нуклеотиды с конца, мы укорачиваем все чтения, а затем убираем те из них, которые в результате этой процедуры стали слишком короткими. Благодаря этому, мы добились пристойного качества на концах прочтений и уменьшили разброс по качеству, начиная примерно с 60 нуклеотида (просто за счёт уменьшения количества длинных, некачественных в конце ридов).
Часть 2: картирование чтений
Выдача программы hisat2 выглядит следующим образом:
11933 reads; of these:
11933 (100.00%) were unpaired; of these:
117 (0.98%) aligned 0 times
11112 (93.12%) aligned exactly 1 time
704 (5.90%) aligned >1 times
99.02% overall alignment rate
Иными словами, из 119333 на хромосому однозначно откартировалось 11112 чтений. Ещё 704 откартировались неоднозначно и 117 вообще никуда не откартировались.
Часть 3: aнализ SNP
Рассмотрим несколько строк файла vcf.
Позиция Референс Что мы наблюдаем Качество Глубина покрытия chr13 25517089 TCCC TCC 217.468 103 chr13 20006620 C T 221.999 19 chr13 25518199 A G 5.46383 1
В верхней строке описана делеция, а в двух других - замены. Посдедний SNP приведён как иллюстрация ошибки: низкое качество и глубина в одно прочтение говорят в пользу того, что это не настоящий полиморфизм, а просто ошибка секвенирования, тогда как, например, делеция из первой строки, по всей видимости, действительно имеет место (причём отмечено, что она встречается примерно в половине прочтений, IDV=51;IMF=0.495146, что говорит о гетерозиготности в этом месте).
Всего в файле vcf 163 снипа (115 transitions, 48 transversions) и 15 инделей. Transition - мутация, при которой пуриновое основание переходит в пуриновое или пиримидиновое в пиримидиновое, transversion - наоборот.
На какие категории делит snp база данных refseq в annovar? Сколько snp у Вас попало в каждую группу?
exonic(9), intronic(90), intergenic(2), upstream(1), downstream(1), UTR5(1), ncRNA_exonic(12), ncRNA_intronic(61)
В какие гены попали Ваши snp?
TPTE2P1, COL4A1(исходя из выдачи refgene, учитываются только полиморфизмы, попавшие в экзоны)
К каким нуклеотидным и аминокислотным заменам привели snp?
6 синонимичных, 3 несинонимичных(refgene)
Сколько snp имеет rs?
140(по базе данных dbsnp)
Что Вы можете сказать о частоте найденных snp?
Частоту можно узнать из выдачи 1000genomes
Что Вы можете сказать о клинической аннотации snp?
В выдаче gwas:
Name=Obesity-related traits chr13 25533831 25533831 A C het 225.009 78 Name=Cardiac hypertrophy chr13 50080847 50080847 A G hom 221.999 45 Name=Arterial stiffness chr13 110818598 110818598 T G het 212.009 49
То есть найдены SNP, ассоциированные с ожирением, сердечной гипертрофией и артериальной ригидностью.
© Быкова Даша, 2018