Анализ транскриптомов
| Программа | Что делает |
| fastqc chr13.1.fastq | Анализ качества чтений |
| hisat2 -x ../chr13 -U chr13.1.fastq -S chr13.1.sam --nosoftclip &> hisat2_output | Кладёт прочтения на референс (запрещаем подрезание концов, а вот разрывать прочтения, в отличие от предыдущего практикума, не запрещаем, так как помним, что имеем дело с транскриптомом) |
| samtools view chr13.1.sam -b -o chr13.1.bam | Преобразование полученного на предыдущем шаге sam - файла в бинарный bam - файл |
| samtols sort chr13.1.bam chr13.1.sorted | Сортировка по координате прочтения (относительно референса) |
| samtools index chr13.1.sorted.bam | Индексирование bam - файла |
| htseq-count -s no -f bam chr13.1.bam -i gene_id -o htseq-count.out.genes /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseq-count.out | Считает чтения, упавшие на разные участки референса |
1. Подготовка чтений
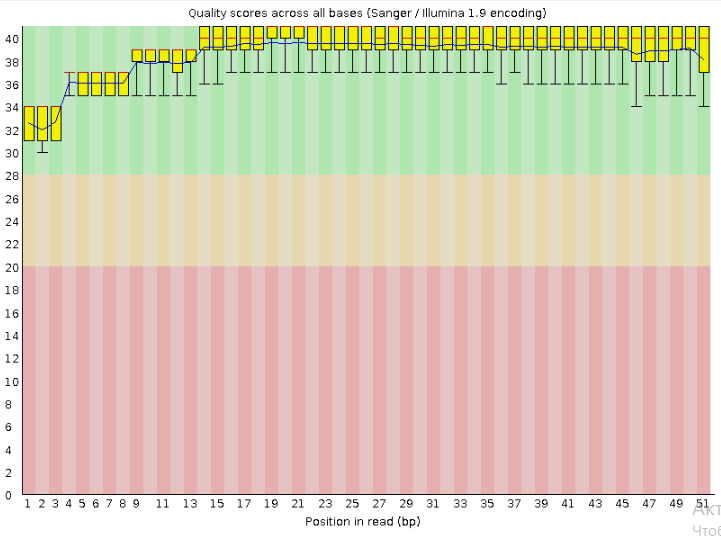
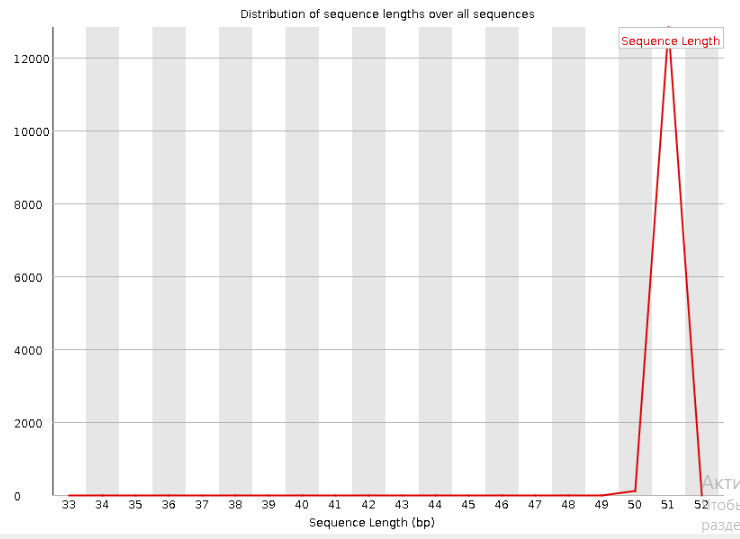
Программа fastqc показала, что чтения, в среднем, имеют неплохое качество. Всего чтений 12985, их длина колеблется от 34 до 51 (как видно из картинки ниже, подавляющее большинство чтений имеют длину около 51). Видимо, из-за меньшей длины по сравнению с прочтениями предыдущего практикума практически не наблюдается падения качества к концу. Было принято решение не использовать программу trimmomatic за ненадобностью.
2. Картирование чтений
При картировании был убран параметр --no-spliced-alignment, так как в транскриптоме после сплайсинга вполне могут оказаться рядом участки, разделённые в геноме интронами, так что нет никакого смысла запрещать программе разрывать прочтение, когда она выкладывает его на референс.
3. Анализ выравнивания
Программа hisat2 на этот раз выдала вот что:
12985 reads; of these:
12985 (100.00%) were unpaired; of these:
121 (0.93%) aligned 0 times
12864 (99.07%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.07% overall alignment rate
Из 12985 прочтений 121 не нашли себе места на референсной хромосоме, а все остальные легли ровно 1 раз.
4. Подсчёт чтений
В программе htseq-count были использованы опции -s, -f, -i. -s отвечает за цепь, из которой пришло прочтение, в данном случае цепь мы заранее не знаем. -f - это формат файла, подающегося на вход программе, по умолчанию - sam, в нашем случае - bam. -i - это 'feature identificator' (видимо, то, по чему мы распознаём, куда легло наше прочтение), в данном случае нам подходит gene_id. Опция -m не была использована, так как в документации к программе при анализе транскриптомов рекомендуется оставить её по умолчанию union (эта опция отвечает за то, как программа будет интерпретировать положение прочтения относительно референсных генов - какое положение считать перекрыванием, а какое - нет).
5. Анализ результатов
Уже отсортированная, с убранными повторяющимися строчками выдача программы htseq-count выглядит вот так:
XF:Z:ENSG00000133112.12 XF:Z:ENSG00000253051.1 XF:Z:__ambiguous[ENSG00000133112.12+ENSG00000199477.1] XF:Z:__ambiguous[ENSG00000273149.1+ENSG00000133112.12] XF:Z:__no_feature XF:Z:__not_aligned
Таким образом, нашлось всего 4 гена, в которые легли наши чтения.
Найдём упомянутые гены в EMBL-EBI
gene_id Координаты(chr13) Описание из базы данных ENSG00000133112 45333471..45341370 Translationally-controlled tumor protein, TPT1, involved in calcium binding and microtubule stabilization ENSG00000199477 45337480..45337609 small nucleolar RNA, H/ACA box 31 ENSG00000253051 45336314..45336447 small nucleolar RNA, H/ACA box 31B ENSG00000273149 45340039..45341183 novel transcript, antisense to TPT1
© Быкова Даша, 2018