Задание 1. Предсказание вторичной структуры заданной тРНК
Для предсказания вторичной структуры тРНК сначала была использована программа einverted, в основе работы которой лежит метод поиска инвертированных повторов в последовательности нуклеиновой кислоты. Стандартные настройки этой программы не позволяют найти ни одного такого повтора, так как тРНК слишком короткая, чтобы проходить порог для веса = 50. Порог был опущен до 20, match score увеличен до 5, mismatch score уменьшен до -6. Были перепробованы разные комбинации параметров. Gap penalty было решено оставить равным 12, так как при его уменьшении программа находила длинные неправильные участки комплементарности. Контринтуитивным кажется решение уменьшить mismatch score, однако практика показывает, что так результаты получаются чуть более адекватными. Итоговый вариант выдачи программы, использованный в анализе:
SEQUENCE: Score 28: 8/10 ( 80%) matches, 0 gaps
23 gagcgcaccc 32
|| | |||||
49 ctggagtggg 40
SEQUENCE: Score 24: 6/7 ( 85%) matches, 0 gaps
1 gggcttg 7
|| ||||
63 cctgaac 57
Далее была сделана попытка предсказать вторичную структуру при помощи программы RNAfold, основанной на расчёте минимальной энергии. Использовался web-вариант программы. Подходящая структура была получена со второго раза, когда в Advanced options в Energy Parameters вместо "RNA parameters (Turner model, 2004)" было выбрано "RNA parameters (Andronescu model, 2007)". Модель, использованная в программе по умолчанию, давала совсем другую структуру, мало похожую на тРНК.
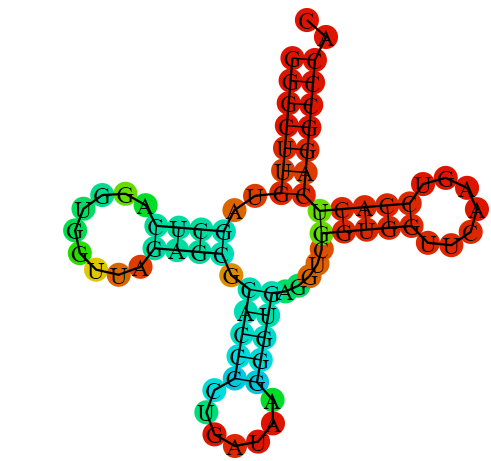
Изображение предсказанной RNAfold вторичной структуры:
Сравнение реальной вторичной структуры тРНК 1ffy и предсказаннной программами einverted и RNAfold
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера (RNAfold) |
| Акцепторный стебель | 5'-1-72-3'; 5'-2-71-3'; 5'-3-70-3'; 5'-4-69-3'; 5'-5-68-3'; 5'-6-67-3'; 5'-7-66-3' Всего 7 пар | einverted сопоставляет нуклеотидам с номерами 1-7 нуклеотиды 63-57, т.е. не находит нужные пары* | Правильно предсказаны все 7 |
| D-стебель | 10-25; 11-24; 12-23; 13-22; Всего 4 пары | einverted не находит пар | Все 4 предсказаны верно |
| Т-стебель | 49-65; 50-64; 51-63; 52-62; 53-61; Всего 5 пар | Нулеотиду с номером 49 поставлен в пару нуклеотид 23 | Все 5 предсказаны верно |
| Антикодоновый стебель | 39-31; 40-30; 41-29; 42-28; 43-27; Всего 5 пар | 27-31 нуклеотидам сопоставляются нуклеотиды 41-45 | Все 5 предсказаны верно |
| Общее число канонических пар нуклеотидов | 20 | 14 | 19 |
Из полученных результатов видно, что для предсказания вторичной структуры более разумно использовать программы, основанные на алгоритме Зукера, чем программы, основанные на поиске инвертированных повторов.
Задание 2. Поиск ДНК-белковых контактов в заданной структуре
Количество контактов разных типов в ДНК-белковом комплексе 1r4o.pdb (количество контактирующих атомов разных типов)
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 1 (10) | 20 (20) | 21 (30) |
| остатками фосфорной кислоты | 4 (11) | 16 (9) | 20 (20) |
| остатками азотистых оснований со стороны большой бороздки | 4 (4) | 9 (10) | 13 (14) |
| остатками азотистых оснований со стороны малой бороздки | 0 | 1 (15) | 1 (15) |
Комментарии к таблице: в задании под контактом подразумевалась пара полярных/неполярных атомов, находящихся на определённом расстоянии. И здесь имеет место некоторая неоднозначность, поскольку в таком случае методом простого пересечения множеств атомов количество контактов определить невозможно. Команды "select within(r,какое-то мн-во атомов ДНК) and (какое-то мн-во атомов белка)" и "select within (r,какое-то мн-во атомов белка) and (какое-то мн-во атомов ДНК)" предсказуемо дают разные результаты, потому что один атом в таком определении может образовывать несколько контактов. В таблицу внесены оба варианта, так что числа - это уже не количество контактов, а количество контактирующих атомов: без скобок - белка, в скобках - ДНК. Число контактов будет равно либо большему из этих чисел, либо чуть больше него (если встретятся атомы ДНК, контактирующие сразу с несколькими белковыми). Несмотря на оговорки, по данной таблице можно сделать какие-то выводы. Например, о том, что наибольший вклад в ДНК-белковое взаимодействие вносят контакты атомов белка с остовом: полярные с остатками фосфорной кислоты и неполярные с сахаром. С атомами большой и малой бороздки белок контакирует примерно поровну.
Упр. 3
Посмотреть изображение к упр.3 можно здесь
Упр.4
а) Как можно видеть на схеме, больше всего контактов (3) с ДНК образует лизин 461 из цепи А.
b) На роль самого важного для распознавания ДНК остатка могут претендовать те, что контактируют с азотистыми основаниями. Наше внимание особенно привлекает Arg466(A), образующий сразу 2 водородные связи с 14 гуанином.
Контакт между [ARG]466:A и [DG]14:C
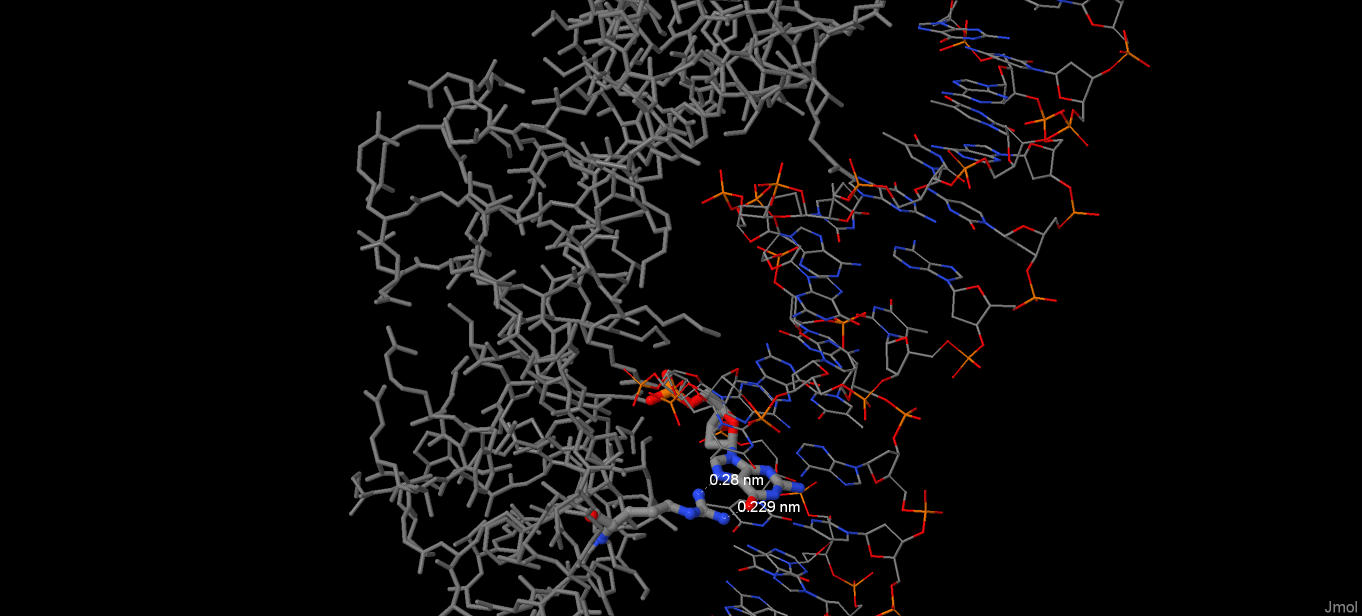
Как видно, аргинин взаимодействует своими аминогруппами с O6 и N7 гуанина. Аналогично поступает и аргинин на другой цепи. Примечательно, что соседние аргинины 489 и 496 при этом контактируют с фосфатной группой.
Контакт между [LYS]461:A и [DG]4:C, а также [LYS]511:A и [DA]3:C
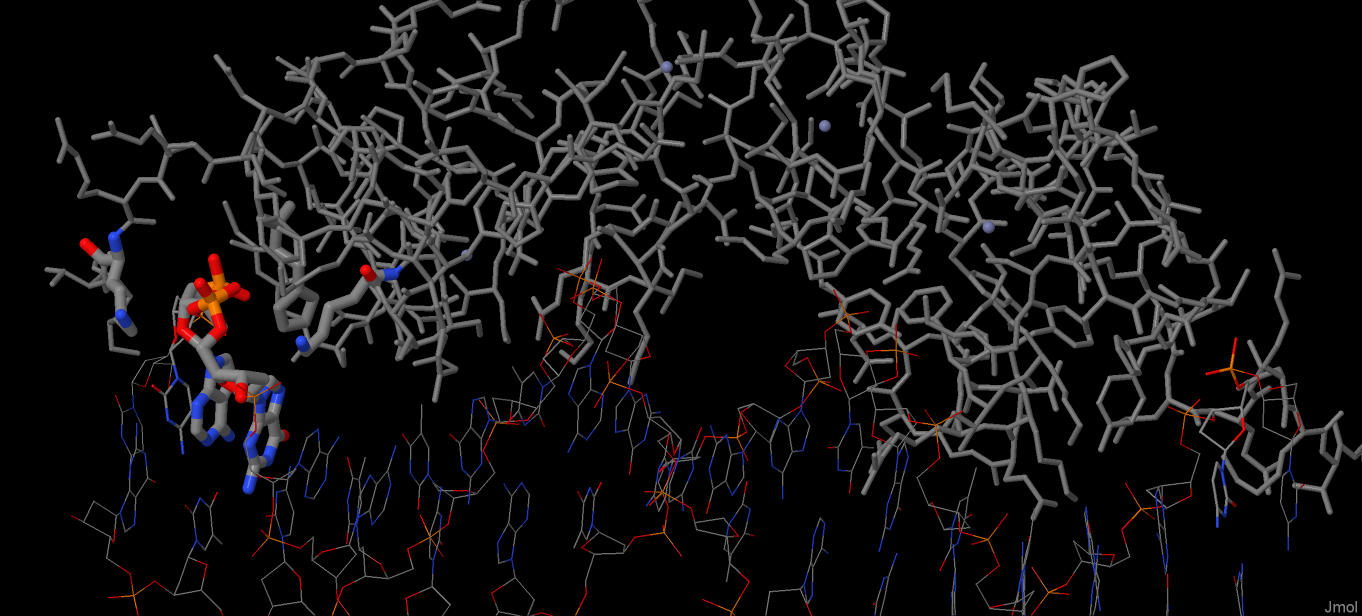
Видимо, здесь имеет место гидрофобное взаимодействие (далековато для чего-то ещё). Лизины как бы обхватывают пурины, а соседние Tyr452 и His451 водородными связями с фосфатными группами укрепляют контакт.
© Быкова Даша, 2018