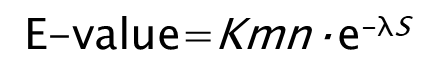
EMBOSS и локальный BLAST
Задание 1
| Номер задания | Задание | Исходные данные | Команды с параметрами | Результаты |
| 1 | Несколько файлов в формате fasta собрать в единый файл | Исходный файл 1 Исходный файл 2 Исходный файл 3 Исходный файл 4 |
seqret @seqlist.txt joined.fasta |
Результат |
| 2 | Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы | Исходный файл |
seqretsplit seq2.fasta -auto |
Результат 1 Результат 2 Результат 3 |
| 3 | Из файла с аннотированной хромосомой в формате gb (из GenBank или RefSeq) или embl (из ENA) вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле | Исходный файл |
seqret @list3.txt 3cds.fasta |
Результат |
| 4 | Транслировать (с первого кодона, то есть в первой рамке) кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода, и положить результат в один fasta файл | Исходный файл |
transeq seq4.fasta -frame 1 -table 0 seq4_translated.fasta |
Результат |
| 5 | Вывести открытые рамки длиной не менее заданной, имеющиеся в данной нуклеотидной последовательности | Исходный файл |
getorf seq5.fasta -minsize 30 seq5_orfs.fasta |
Результат |
| 6 | Перевести выравнивание из формата fasta в формат msf | Исходный файл |
seqret ali6.fasta msf::ali6.msf |
Результат |
| 7 | Выдать в файл число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имена последовательностей и числа) | Исходный файл |
infoalign -refseq 2 -idcount -name -only ali7.fasta ali7_idcount |
Результат |
| 8 | Перевести аннотации особенностей из файла формата gb или embl в табличный формат gff | Исходный файл |
featcopy Pseudomonas_phage_F116.gb pseudomonas_phage_f116.gff |
Результат |
| 9 | Из данного файла с хромосомой в формате gb или embl получить fasta файл с кодирующими последовательностями | Исходный файл |
extractfeat -type CDS Pseudomonas_phage_F116.gb ppf116_cds.fasta |
Результат |
| 10 | Перемешать буквы в данной нуклеотидной последовательности | Исходный файл |
shuffleseq seq10.fasta seq10_shuffled.fasta |
Результат |
Задание 2
Проверить, сколько находок с E-value < 0.1 в среднем находит blastn для случайной последовательности данной длины в данном геноме бактерии.
Скрипт 1 принимает на вход длину случайной последовательности, которую мы будем искать, и геном, в котором будем её искать. Далее скрипт создаёт 100 случайных последовательностей заданной длины, ищет их в заданном геноме при помощи blastn и выдаёт количество находок с pvalue < 0.1. Понятно, что единичный запуск скрипта даёт довольно мало информации. Немного усовершенствуем скрипт и соберём статистику (в качестве рассматриваемого взят геном Bacillus subtilis, NC_000964.3).
Ссылка на таблиицу с результатами
Усовершенствованный скрипт принимает на вход те же параметры, однако проделывает операции первого скрипта 100 раз и записывает результат в файл с указанием длины искомых случайных последовательностей. Вспомним, что e-value вычисляется по следующей формуле (K и лямбда - константы, зависящие от используемой матрицы и штрафа за гэпы, m - длина искомой последовательности, n - размер банка, в нашем случае тоже константа, S - вес):
Или (вес в битах B):
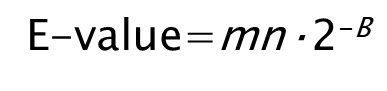
При увеличении длины запроса e-value должно увеличиваться. Кажется, что отсюда следует, что при увеличении длины искомой последовательности должно появляться меньше находок с маленьким e-value. Однако (см. таблицу) этого не происходит. По количеству находок (и среднему, и медиане, и максимальному) мы наблюдаем волнообразное изменение с минимумом при length = 50 и падением при length = 400-500. Возможно, данный факт объясняется тем, что с увеличением длины запроса увеличивается и шанс найти случайно похожий кусок в базе. И пусть в среднем e-value таких находок падает (может быть, доля тех, что имеют e-value < 0.1 и становится меньше), но за счёт увеличения их общего числа количественного падения мы не наблюдаем.
© Быкова Даша, 2018