Сравнение матриц аминокислотных замен
Для оценки качества выравнивания применяются матрицы аминокислотных замен, которые строятся на основе баз данных аминокислотных последовательностей. В подобных матрицах чем больше значение, тем выше вероятность такой замены. Матриц аминокислотных замен существует несколько видов, вот некоторые из них:
BLOSUM62-матрица, созданная на основе базы данных для цитоплазматических белков BLOCKS. Число 62 в названии указывает на величину порога кластеризации, то есть процент похожести, начиная с которого, последовательности кластеризуются и рассматриваются как единое целое, чтобы не сдвигать значения вероятностей.
PHAT-матрица, созданная на основе базы данных для трансмембранных белков. Поскольку трансмембранные и цитоплазматические белки имеют разную специализацию, встречаемость одних и тех же аминокислот в них разная.
Я воссоздал матрицу BLOSUM на основе базы данных BLOCKS, рассчеты этой реконструкции представлены Здесь. В общем, полученная матрица очень похожа на оригинальную BLOSUM62: большинство значений отличаются по абсолютной величине, но сохраняются все соотношения. Вероятно, к отличиям привел тот факт, что оригинальная матрица составлялась давно и с тех пор база данных расширилась, также различия вызваны тем, что при реконструкции я не учитывал B, Z, X и ГЭПы, что, в свою очередь, несколько исказило вероятности. Для примера ниже представлена таблица весов замены лейцина на другие аминокислоты по трем описанным матрицам:
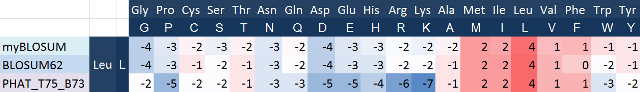
Как видно на таблице, вес замены лейцина на самого себя одинаковый и весьма высокий для всех матриц. Вероятность замены аминокислоты на какую-нибудь другую гораздо меньше, чем самой на себя, что связано с процессивностью ДНК-полимеразы.
Замены на другие гидрофобные аминокислоты с крупным радикалом также мало отличаются и имеют большой вес. Очевидно, такие замены не сильно влияют на структуру белка, поскольку их свойства очень похожи, и вероятность таких замен высока.
Замены на полярные и отрицательно заряженные кислоты, такие как глутамат и глутамин, имеют маленький вес, поскольку сильно отличаются по свойствам от лейцина, что мешает белку принять правильную структуру. Значения для матрицы PHAT несколько ниже, потому что если гидрофобный участок перестанет быть таковым, белок может не встроиться в мембрану и не выполнять свои функции. Особенно маловероятна для трансмембранных белков замена гидрофобного лейцина на положительно заряженные гистидин, аргинин и лизин, так как это в подавляющем большинстве случаев не даст ему функционировать правильно, соответсвенно такие замены по матрице PHAT имеют очень маленькие и сильно отличающиеся от прочих матриц веса.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
По алгоритму Нидлмана-Вунша работает программа needle пакета EMBOSS. Этот алгоритм выравнивает целые последовательности и находит наибольший процент схожести
По алгоритму Смита-Ватермана работает программа water пакета EMBOSS. Этот алгоритм выравнивает куски последовательностей.
По умолчанию для оценки веса выравнивания используется матрица BLOSUM62. В качестве параметров эти программы имеют штраф за первый ГЭП и за последующие (значения по умолчанию 10 и 0,5 соответственно).
Ниже приведены выравнивания сделанные вручную и с использованием обоих алгоритмов для белка pdxt_bacsu и трех коротких мутантов (20 а.к.), полученных с помощью скрипта evolve_protein.pl со следующими параметрами:
Параметр |
Мутант №1_1 |
Мутант №2_1 |
Мутант №3_1 |
Вероятность изменения остатка (моделирующая "ошибку" ДНК-полимеразы) |
0.6 |
0.6 |
0.4 |
Вероятность замены остатка в случае, если данная позиция будет изменена |
0.6 |
0.8 |
0.8 |
Выравнивания
Мутант №1_1 вручную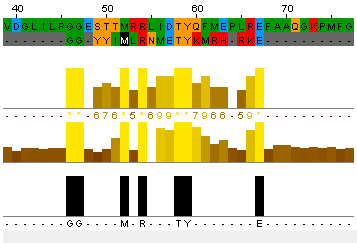
lenght=22
identity=31.8%
similarity=59%
water №1_1
# Length: 9
# Identity: 4/9 (44.4%)
# Similarity: 7/9 (77.8%)
# Gaps: 0/9 ( 0.0%)
# Score: 21.0
#=======================================
PDXT_BACSU 52 MRRLIDTYQ 60
|.|.::||:
generations=1 6 MLRNMETYK 14
needle №1_1
# Length: 196
# Identity: 7/196 ( 3.6%)
# Similarity: 11/196 ( 5.6%)
# Gaps: 176/196 (89.8%)
# Score: 18.0
#=======================================
PDXT_BACSU 1 MLTIGVLGLQGAVREHIHAIEACGAAGLVVKRPEQLNEVDGLILPGGEST 50
|...
generations=1 1 ----------------------------------------------GGYY 4
PDXT_BACSU 51 TMRRLIDTYQFMEPLREFAAQGKPMFGTCAGLIILAKEIAGSDNPHLGLL 100
.|.|.::||: |...:|
generations=1 5 IMLRNMETYK-MRHRKE--------------------------------- 20
PDXT_BACSU 101 NVVVERNSFGRQVDSFEADLTIKGLDEPFTGVFIRAPHILEAGENVEVLS 150
generations=1 20 -------------------------------------------------- 20
PDXT_BACSU 151 EHNGRIVAAKQGQFLGCSFHPELTEDHRVTQLFVEMVEEYKQKALV 196
generations=1 20 ---------------------------------------------- 20
Мутант №1_2 вручную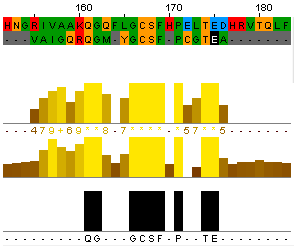
lenght=22
identity=40.9%
similarity=63.6%
water №1_2
# Length: 13
# Identity: 6/13 (46.2%)
# Similarity: 9/13 (69.2%)
# Gaps: 1/13 ( 7.7%)
# Score: 33.0
#=======================================
PDXT_BACSU 157 VAAKQGQFLGCSF 169
:..:||.: ||||
generations=1 3 IGQRQGMY-GCSF 14
needle №1_2
# Length: 196
# Identity: 9/196 ( 4.6%)
# Similarity: 12/196 ( 6.1%)
# Gaps: 176/196 (89.8%)
# Score: 26.0
#=======================================
PDXT_BACSU 1 MLTIGVLGLQGAVREHIHAIEACGAAGLVVKRPEQLNEVDGLILPGGEST 50
generations=1 0 -------------------------------------------------- 0
PDXT_BACSU 51 TMRRLIDTYQFMEPLREFAAQGKPMFGTCAGLIILAKEIAGSDNPHLGLL 100
generations=1 0 -------------------------------------------------- 0
PDXT_BACSU 101 NVVVERNSFGRQVDSFEADLTIKGLDEPFTGVFIRAPHILEAGENVEVLS 150
generations=1 0 -------------------------------------------------- 0
PDXT_BACSU 151 EHNGRIVAAKQGQFLGCSFHPELTEDHRVTQLFVEMVEEYKQKALV 196
..:..:||.: |||| |..||.
generations=1 1 ----VAIGQRQGMY-GCSF-PCGTEA-------------------- 20
Мутант №1_3 вручную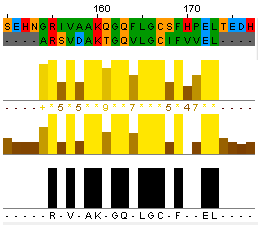
lenght=20
identity=60%
similarity=80%
water №1_3
# Length: 19
# Identity: 12/19 (63.2%)
# Similarity: 12/19 (63.2%)
# Gaps: 0/19 ( 0.0%)
# Score: 50.0
#=======================================
PDXT_BACSU 155 RIVAAKQGQFLGCSFHPEL 173
|.|.||.||.|||.|..||
generations=1 2 RSVDAKTGQVLGCIFVVEL 20
needle №1_3
# Length: 196
# Identity: 12/196 ( 6.1%)
# Similarity: 12/196 ( 6.1%)
# Gaps: 176/196 (89.8%)
# Score: 50.0
#=======================================
PDXT_BACSU 1 MLTIGVLGLQGAVREHIHAIEACGAAGLVVKRPEQLNEVDGLILPGGEST 50
generations=1 0 -------------------------------------------------- 0
PDXT_BACSU 51 TMRRLIDTYQFMEPLREFAAQGKPMFGTCAGLIILAKEIAGSDNPHLGLL 100
generations=1 0 -------------------------------------------------- 0
PDXT_BACSU 101 NVVVERNSFGRQVDSFEADLTIKGLDEPFTGVFIRAPHILEAGENVEVLS 150
generations=1 0 -------------------------------------------------- 0
PDXT_BACSU 151 EHNGRIVAAKQGQFLGCSFHPELTEDHRVTQLFVEMVEEYKQKALV 196
.|.|.||.||.|||.|..||
generations=1 1 ---ARSVDAKTGQVLGCIFVVEL----------------------- 20
Значения identity и similarity значительно отличаются при выравнивании по алгоритму Нидлмана, поскольку при этом выравнивается вся последовательность и учитывается множество ГЭПов. Значения для ручного выравнивания и выравнивания по алгоритму Ватермана хорошо сопоставимы, однако программой чуть выше, вероятно, ввиду того что она рассчитывает их для куска меньшей длины, а при ручном способе не учитываются многие факторы такие, как штрафы за ГЭПы.
Значения identity и similarity значительно отличаются при выравнивании по алгоритму Нидлмана, поскольку при этом выравнивается вся последовательность и учитывается множество ГЭПов. Значения для ручного выравнивания и выравнивания по алгоритму Ватермана хорошо сопоставимы, однако программой чуть выше, вероятно, ввиду того что она рассчитывает их для куска меньшей длины, а при ручном способе не учитываются многие факторы такие, как штрафы за ГЭПы.