Анализ трехмерных структур
d1: Определение вторичной структуры
С помощью программы Stride была определена вторичная структура белка 3bmx. Выдача доступна по ссылке. Первые с N-конца две альфа-спирали и два бета-тяжа были сопоставлены с записью в PDB файле. Если учесть различную нумерацию остатков, границы альфа-спиралей и первого бета-тяжа полностью совпадают. Второй бета-тяж в PDB файле длиннее на один остаток с С-конца.
С помошью сервиса SheeP были построены карты бета-листов. Ниже приведена карта самого большого из них и его положение в белке.
.PNG)
Рис.1. Один из бета-листов белка 3bmx. Сверху представлена карта бета-листа, ниже его положение в структуре.
.PNG)
Рис.2. Рассматриваемый бета-лист отдельно. Белым выделен гребень, соответствующий третьему столбцу.
При помощи сервиса Proton c использованием Original Stride была получена схема водородных связей в данном бета-листе. На схеме также отмечены пептидные связи.
d2: Совмещение структур
С помощью PDBeFold с настройками по умолчанию были найдены структурные гомологи белка 3bmx. PDB файл 3bmx содержит две идентичные цепи: A и B. Для поискового запроса использовалась только цепь А.Из 38 полученных хитов были выбраны 4 наиболее репрезентативные: 4gyk:B, 3wlt:A, 3sql:A и 3rrx:A.
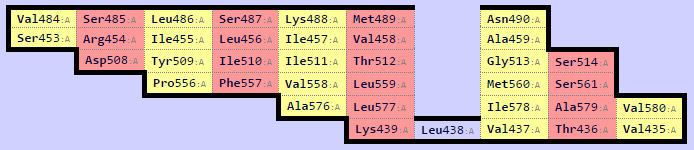
Было получено структурное выравнивание самого белка и выбранных гомологов. Информация о выравнивании сохранена в текстовом файле. Само выравнивание для дальнейшей работы сохранено в формате fasta Ниже представлена анимация полученного совмещения структур.
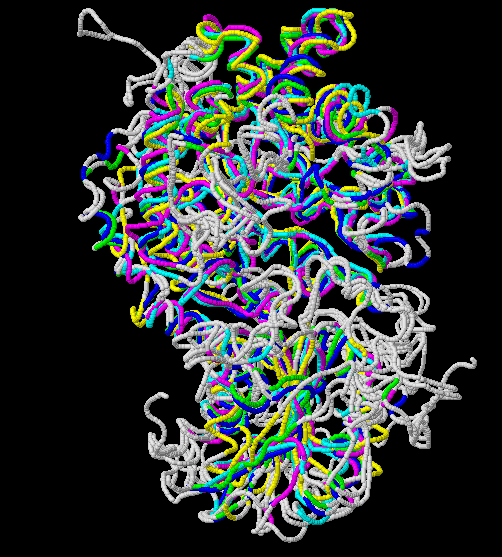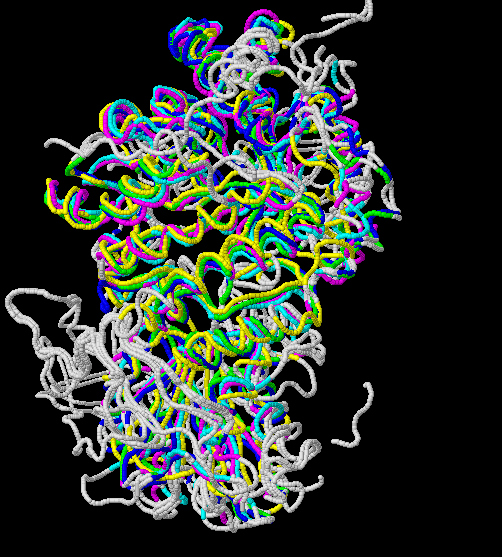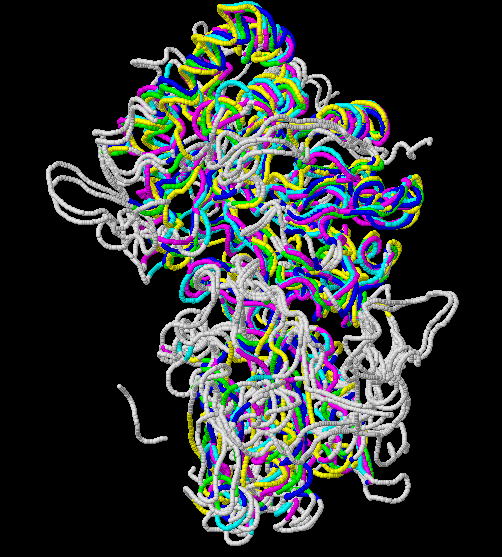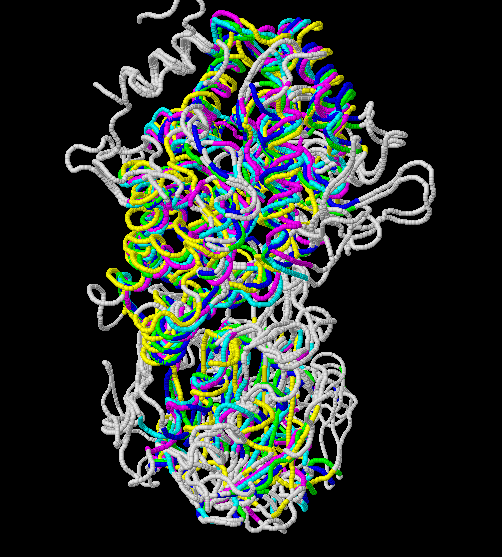
Рис.3. Совмещение, полученное в результате структурного выравнивания. Цветом обозначены участки, выравненнные для всех последовательностей. Структура 3bmx:A покрашена в зеленый, 4gyk:B - синий, 3wlt:A - голубой, 3sql:A -желтый и 3rrx:A - розовый соответственно.
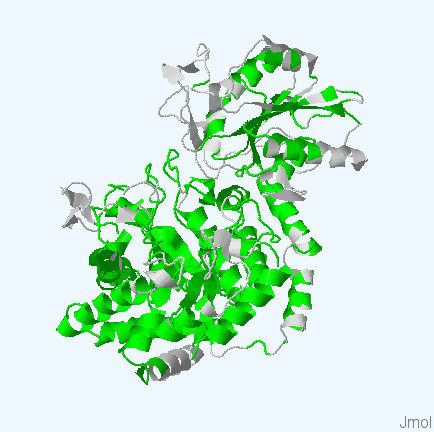
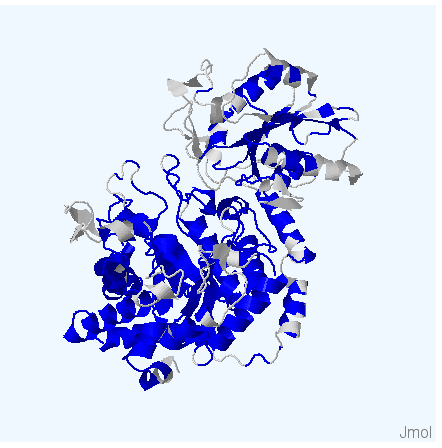
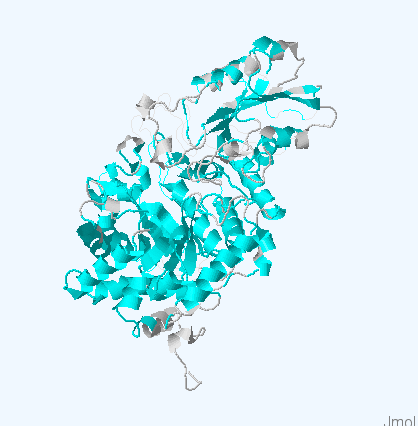
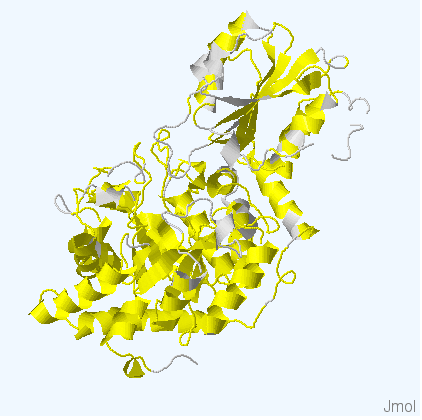
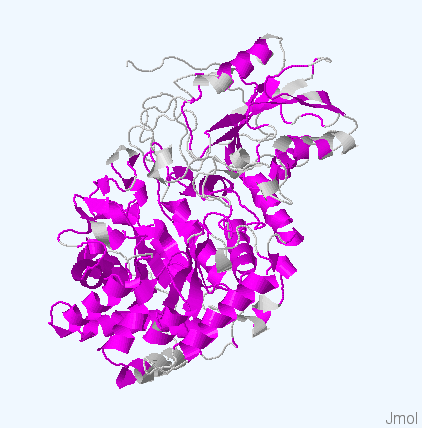
Рис.4. Структуры белков используемых для работы. Цветом обозначены участки, выравненнные для всех последовательностей. Структура 3bmx:A покрашена в зеленый, 4gyk:B - синий, 3wlt:A - голубой, 3sql:A -желтый и 3rrx:A - розовый соответственно.
Полученное выравнивание было окрашено в соответствии с цветовой схемой Clustal с порогом консервативности 30% при помощи программы JalView. С результатом можно ознакомиться по ссылке.
Множественное выравние рассматриваемых белков по последовательности было построено с помощью сервиса Muscle в той же программе. С результатом можно ознакомиться по ссылке. Окраска аналогичная.
Было найдено одно отличие между структурным и выравниванием по последовательности. Так, остаток метионина, имеющий номер 348 в структуре 3bmx, не выровнен в структурном выравнивании, хотя находится на аналогичных местах в трех из пяти последовательностей. На совмещении структур видно, что положение этого остатка в пространстве для разных белков совпадает. В случае выравнивания по последовательности, этот остаток оказывается выровнен.В целом, два выравнивания оказались весьма сходны, хотя, при детальном сравнении, можно заметить, что выравнивание по последовательности в данном случае оказалось немного лучше структурного. Например, структурное выравнивание зачастую пропускает явно гомологичные остатки. Огрехи структурного выравнивания также заметны на представленной выше анимации. Тут в местах, в которых структуры достоверно совпадают, присутствуют неокрашенные пятна.


Рис.5. Отличие состояния остатка метионина при структурном и по последовательности. Участки выравниваний слева - структурное, справа - программой Muscle.
Рис.6. Совмещение структур. Выделен спорный остаток метионина.
С помощью PDBeFold также был проведен поиск структурных гомологов домена A:203-315 аконитазы свиньи 1b0m. Результат поиска сохранен в текстовом файле.
Как ни странно, сам домен, как и исходный белок в выдаче отсутствуют. Вероятно, это вызвано небольшой длиной участка, служившего поисковым запросом.По умолчанию в PDBeFold стоит высокий уровень порога сходства(70%). Короткий поисковой запрос не может обеспечить покрытия, необходимого для достижения порога, поэтому исходный белок в выдаче отсутствует.
Для дальнейшей работы был выбран белок с идентификатором 1OGA. С помощью PyMol были получены PDB структуры константного домена T-клеточного рецептора из цепочки альфа(1oga:D 118-202) и цепи бета (1oga:E 119-245). Координаты доменов получены из базы SCOP.
С помощью сервиса SheeP были получены карты бета-листов для данных цепочек. Они представлены на изображении ниже. В цепи бета оказалось два бета-листа. Для дальнейшей работы использовался самый большой. Кроме того, при визуальном сравнении цепей альфа и бета структура большего бета-листа цепи бета больше соответствует бета-листу цепи альфа.
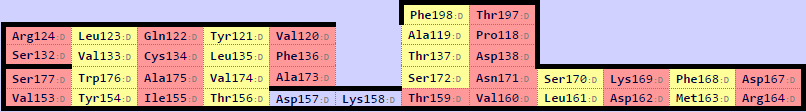
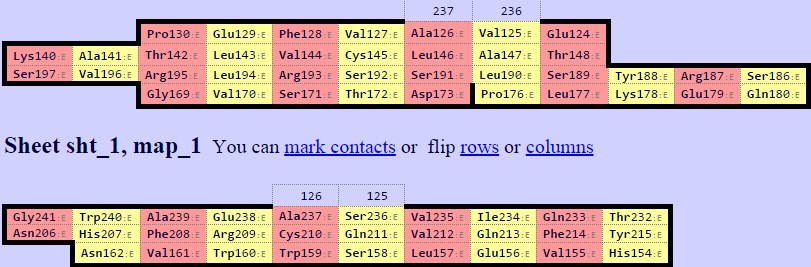
Рис.7. Карты бета-листов.Верхняя для цепи альфа, две нижние для цепи бета.Карты листов находятся в соответствующей друг другу ориентации.
Домены были совмещены в PyMol с помощью скрипта. Выравнивание осуществлялось на основании следующих соображений:остатки цистеина внутри листа должны быть веровнены по умолчанию, также выровненными считались соседние с ним остатки из того же и двух соседних гребней.
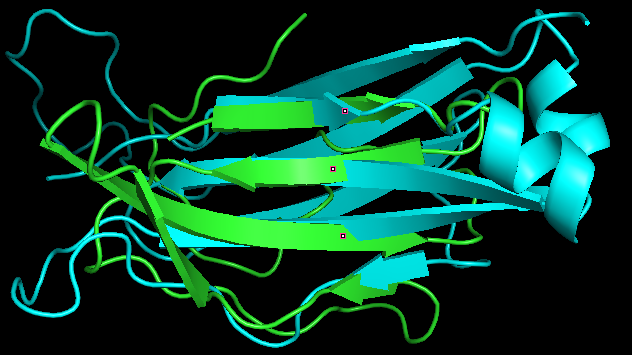
Рис.8. Совмещение бета-листов альфа и бета цепочек константного домена T клеточного домена из структуры 1oga. Зеленой изображена альфа цепочка, бета обозначена голобым.
Как видно на рисунке, топологии бета листов альфа и бета цепочек в некоторой степени совпадают. Так, по крайней мере, совпадают направления и изгиб тяжей. Однако, имеются серьезные отличия. Например, один из тяжей альфа цепи не имеет аналога в бета цепи. Также в бета цепочке имеется короткая альфа-спираль между двумя соседними тяжами, отсутствующая в альфа цепи. Сессия PyMol сохранена в файл.
d3: Нахождение гидрофобных bов
С помощью сервиса CluD был осуществлен поиск гидрофобных bов в структуре 3bmx. Методом проб и ошибок было установлено, что оптимальными параметрами при этом являются порог расстояния 4,5 ангстрема и порог размера 5 атомов. При большем пороге расстояния поиск выдает чрезмерно большие bы, которые не отражают действительность. При меньшем пороге расстояния bы просто разбиваются на множество маленьких..PNG)
Рис.9. Гидрофобные bы в структуре 3bmx. Как видно на картинке, несколько небольших bов объединяются в большие гидрофобные области. Для обоих цепей эти области расположены в ядре двух структурных доменов, различимых на глаз. Оба домена представляют собой глобулы укладки альфа/бета. Гидрофобные области соответствуют ядрам этих глобул. В случае одного домена в центре глобулы лежит бета лист, в случае второго бета-бочонок. Гидрофобные области оказываются в середине бета-бочонка и в месте контакта бета-структур с окружающими их альфа-спиралями.
Далее с помощью того же сервиса был произведен поиск гидрофобных ядер в структуре 2nv2. Поиск производился с теми же параметрами, что и ранее. В этом случае внимание было сосредоточено на bах, расположеных в месте контакта различных мономеров. Так в области контакта цепей А и В обнаружилось 5 гидрофобных bов.
.PNG)
Рис.10. Гидрофобные bы в области контакта цепей А и В структуры 3bmx.
d4: Построение поверхности, раскраска участка поверхности: pymol
Для работы использовалась структура комплекса пуринового репрессора 1PNR с ДНК. PDB файл содержит только один мономер, хотя белок в клетке существует в виде димера. Для восстановления второго мономера в PyMol использовалась следующая команда:symexp sym, 1npr, all, 2.
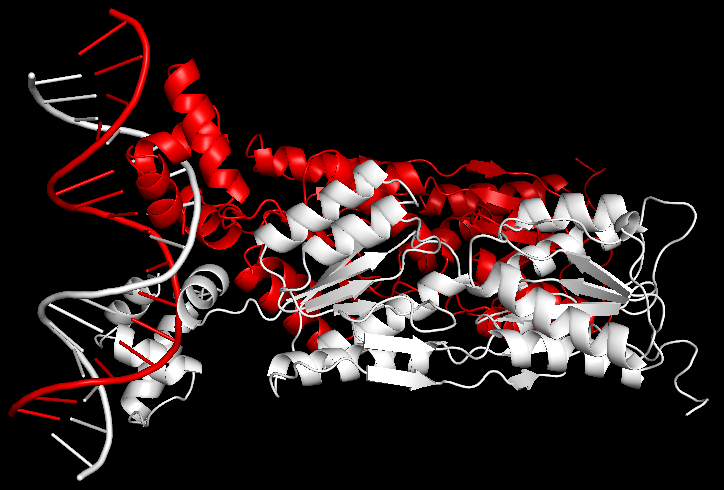
Рис.11. Комплекс димера пуринового репрессора с ДНК 1PNR.
Затем была построена поверхность контакта двух мономеров. Для выделения области контакта использовалась команда: select contact1, /1npr//A near_to 5 of /sym02000000//A.
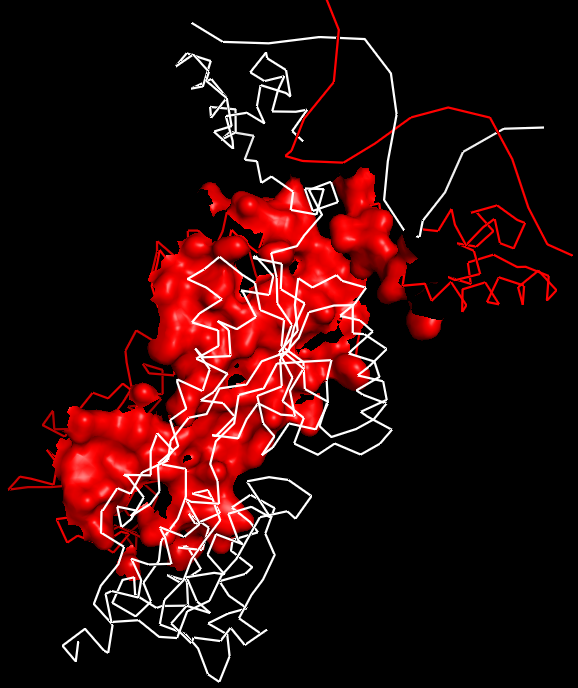
Рис.12. Поверхность контакта мономера белка с симметричным мономером на фоне остовной (ribbon) модели мономеров.
Аналогичным путем были построены поверхности белка в области контакта с ДНК и наоборот, ДНК с белком.
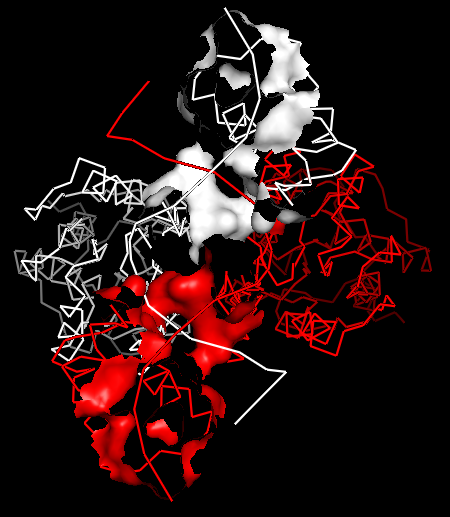
Рис.13. Поверхность контакта димера белков с двойной спиралью ДНК на фоне остовной модели части белка, вовлечённой в контакт.
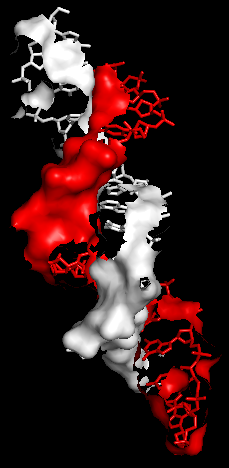
Рис.14. Поверхность контакта ДНК с димером белков на фоне проволочной (sticks) модели двойной спирали.
С помощью сервиса CluD были обнаружены гидрофобные bы в области контакта мономеров. Всего было найдено три подходящих bа. Поиск осуществлялся с порогом по расстоянию в 4,5 ангстрем и порогом по размеру в 10 атомов. Для поискового запроса использовался PDB-файл, содержащий димер.
В выдаче CluD в виде текстового файла были найдены номера атомов, образующих bы. Затем в PyMol эти атомы обозначены на поверхности контакта мономеров.
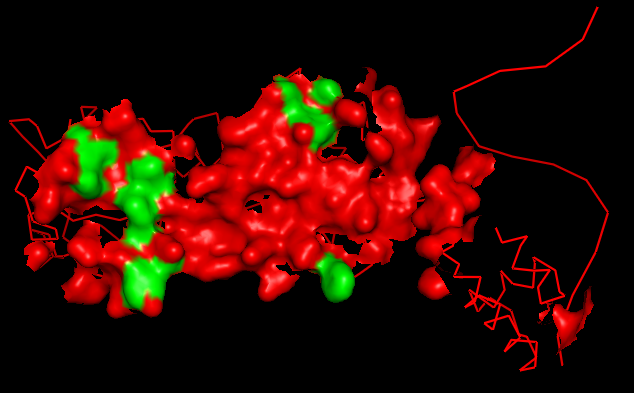
Рис.15. Поверхность контакта мономера белка с симметричным мономером на фоне остовной (ribbon) модели мономеров. Атомы, образующие гидрофобные bы, выделены зеленым цветом.
d5: Сравнение доменов SCOP/SCOPe, ECOD, CATH и Pfam
С помощью различных баз данных был осуществлен поиск доменов белка бета-гексаминидазы, трехмерная модель которой имеет идентификатор 3ВМХ. Результаты занесены в таблицу.
| База данных | ID домена | Границы | Описание |
| SCOPe 2.05 | - | ||
| CATH | 3bmxA01 | 26-419 | TIM бочка |
| 3bmxA02 | - | - | |
| Pfam | Glyco_hydro_3 | 43-392 | Гликозид гидролаза семейство 3, N-концевой домен |
| Glyco_hydro_3_C | 435-636 | Гликозид гидролаза семейство 3, С-концевой домен | |
| ECOD | e3bmxA1 | 26-434 | TIM бочка |
| e3bmxA2 | 435-642 | Бета-D-глюкан экзогидролаза, C-концевой домен | |
Белок содержит два домена. Оба домена нашлись во всех рассматриваемых базах данных, за исключением SCOPe. Это может быть вызвано тем, что модель 3ВМХ относительно новая (2007), а SCOPe перестала активно развиваться. В базе CATH отсутствуют координаты и описание второго (С-концевого) домена. Однако сервис распознает этот домен, хотя и сообщает, что информация о нем в процессе обработки. Границы первоого домена практически совпадают в CATH и ECOD. Границы обоих доменов оказываются значительно урезаны в случае Pfam по сравнению с другими базами данных. Возможно, у Pfam более строгие правила, определяющие домены. Также наблюдаемая ситуация может быть вызвана тем обстоятельством, что Pfam, в отличие от остальных, содержит эволюционные домены, а не структурные. В целом, результаты поиска по различным базам не противоречат друг другу.
d6: Использование сайта PDB
На сайте PDB с помощью Advanced Search (Experimental Method: ELECTRON MICROSCOPY) были найдены все белки, структуры которых определены при помощи метода электронной микроскопии. Их последовательности представлены в виде одного fasta-файла.
Список структурных гомологов 3bmx по результатам жесткого выравнивания с помощью PDBeFold представлен в файле. Список структурных гомологов по результатам гибкого выравнивания jFATCAT доступен по ссылке. Гомологов по результатам PDBeFold значительно меньше, чем по результатам jFATCAT(38 против 284). В целом, jFATCAT находит более эволюционно далеких гомологов. Кроме того, значения rmsd для одних и тех же хитов различается при различных выравниваниях.