Выдача программы:
SP score: 0.78
CS score: 0.55
avg_SPdist score: 0.88
*SP score - процент гомологий в эталонном выравнивании, который появляется в предполагаемом выравнивании
(доля одинаково выровненных пар остатков в тестовом и эталонном выравниваниях),CS score - доля одинаково выровненных позиций,
avg_SPdist - похожа на оценку SP, но включает информацию о расстоянии между парами несовмещенных остатков (Для каждой выровненной
пары остатков в эталонном выравнивании теперь определяется расстояние между соответствующими двумя остатками в выравнивании запроса;
в качестве конечной оценки SPdist рассчитывается среднее расстояние несоответствия по всем выровненным парам в эталонном выравнивании).
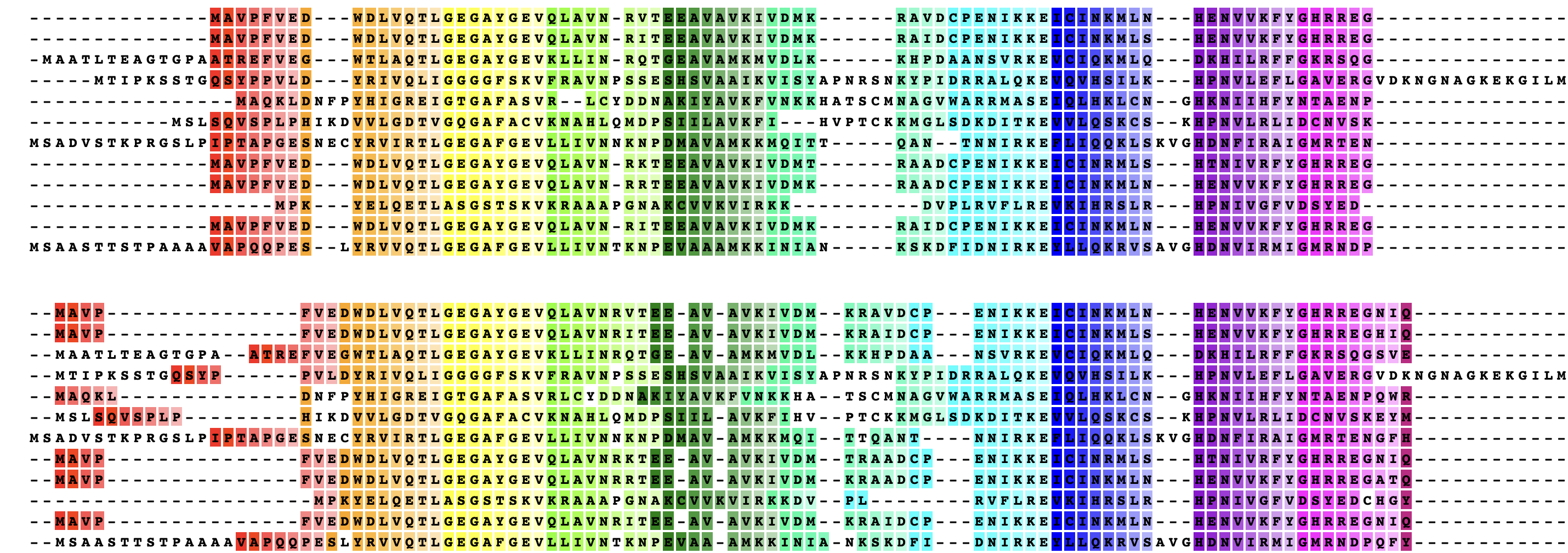
Ниже приведён начальный фрагмент сравнения выравниваний программами Clustal и
Muscle. Можно заметить, что выравнивания совпали на позициях (привожу лишь несколько примеров)
26-41, 74-92, и не совпали на позициях 1-15, 46-49 (высчитано вручную).
Выдача программы:
SP score: 0.85
CS score: 0.69
avg_SPdist score: 0.92
*SP score - процент гомологий в эталонном выравнивании, который появляется в предполагаемом выравнивании
(доля одинаково выровненных пар остатков в тестовом и эталонном выравниваниях),CS score - доля одинаково выровненных позиций,
avg_SPdist - похожа на оценку SP, но включает информацию о расстоянии между парами несовмещенных остатков (Для каждой выровненной
пары остатков в эталонном выравнивании теперь определяется расстояние между соответствующими двумя остатками в выравнивании запроса;
в качестве конечной оценки SPdist рассчитывается среднее расстояние несоответствия по всем выровненным парам в эталонном выравнивании).
Ниже приведён начальный фрагмент сравнения выравниваний программами MSAprobs и
MAFFT.
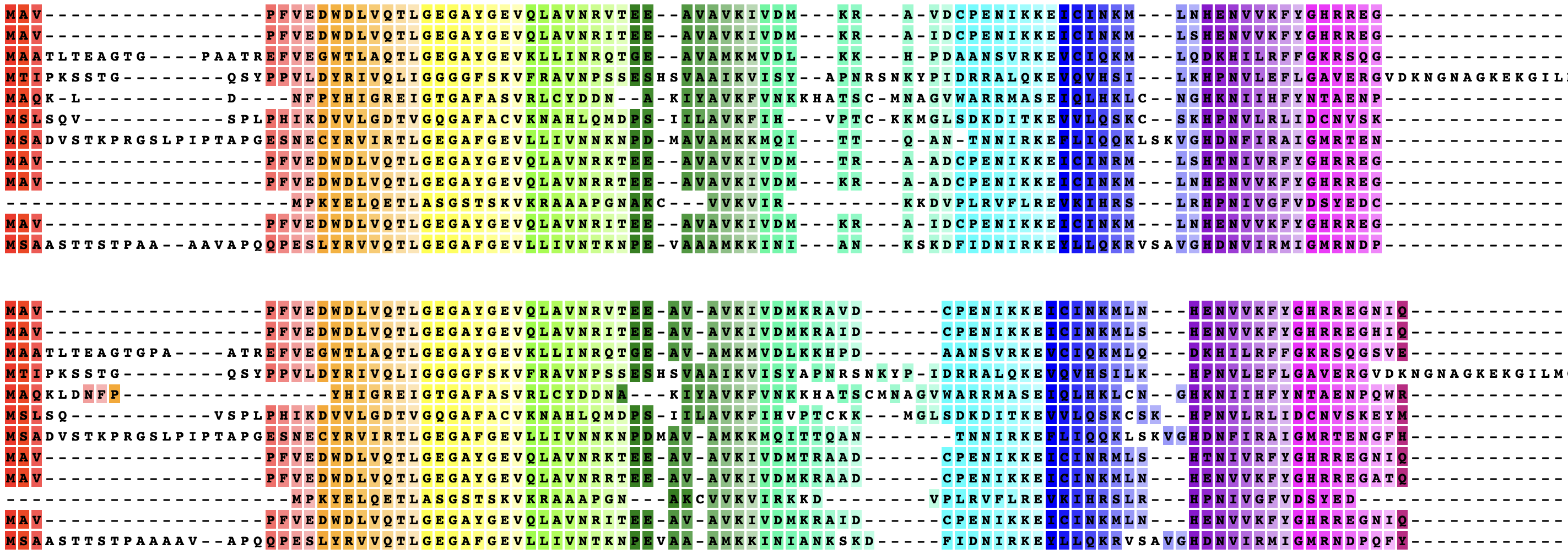
Аналогично (приведу несколько примеров), совпало: 1-4, 19-22, не совпало: 5-9, 23-25.
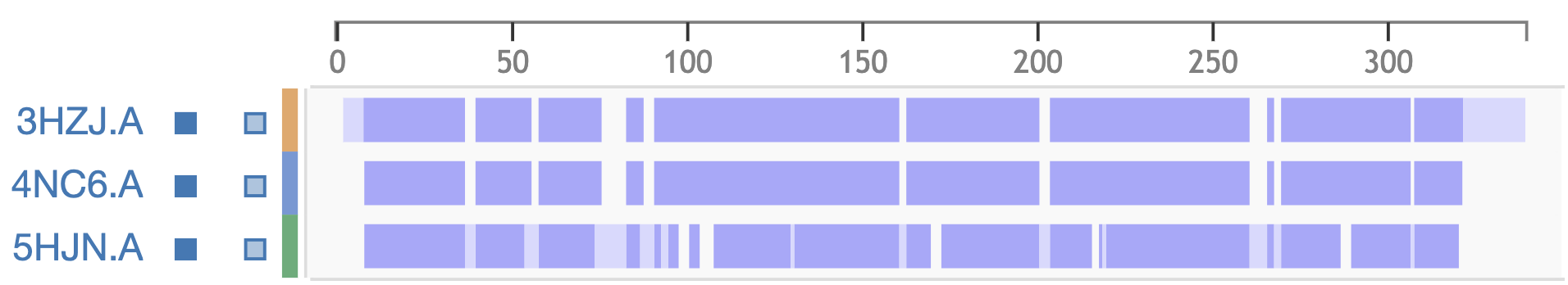
Для выполнения этого задания я выбрала семейство RabGAP-TBC (PF00566) и 3 белка их него (далее указаны PDB ID): 3HZJ, 4NC6, 5HJN. Выравнивание было произведено в PyMOL с помощью команды align.
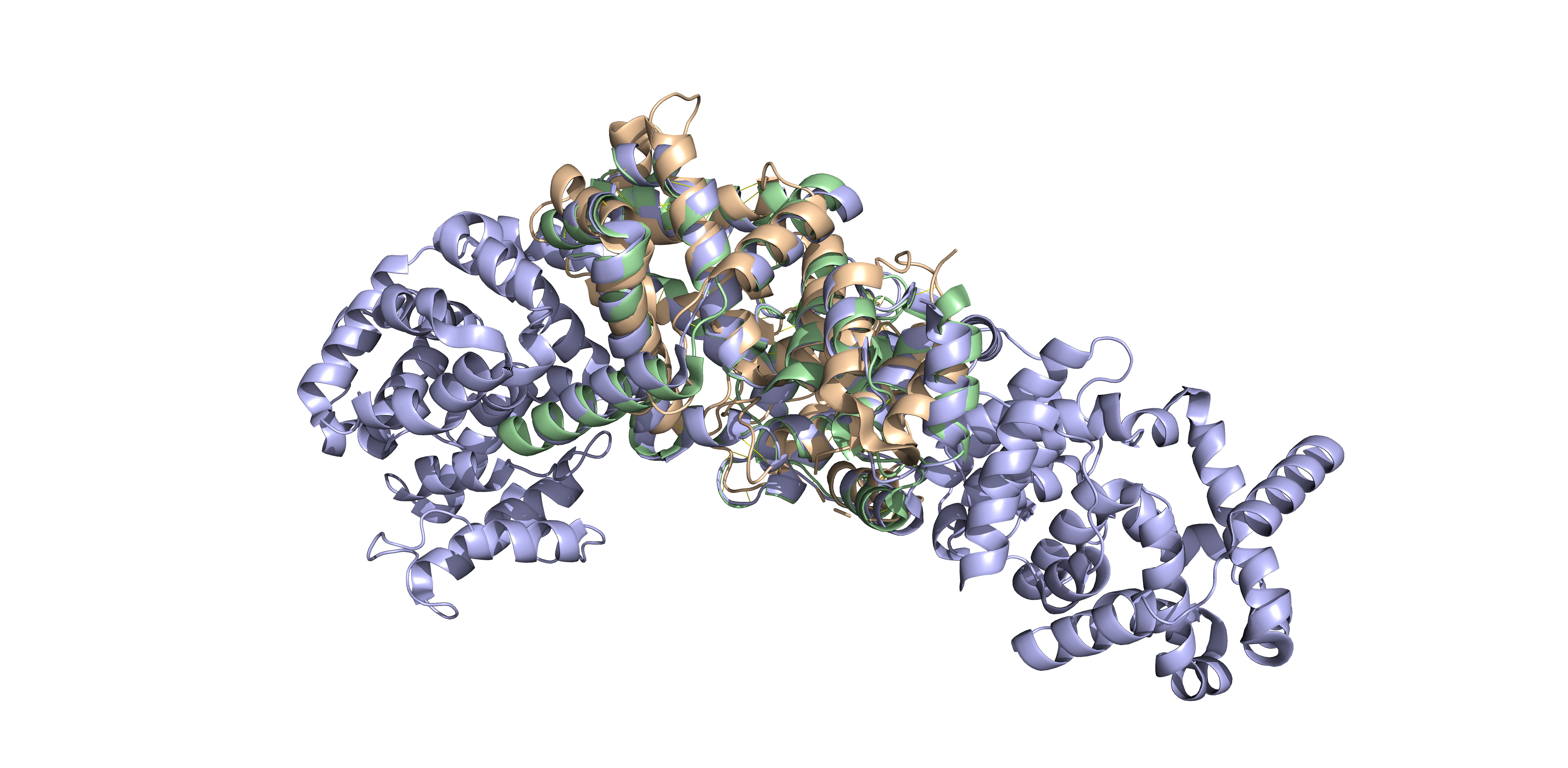
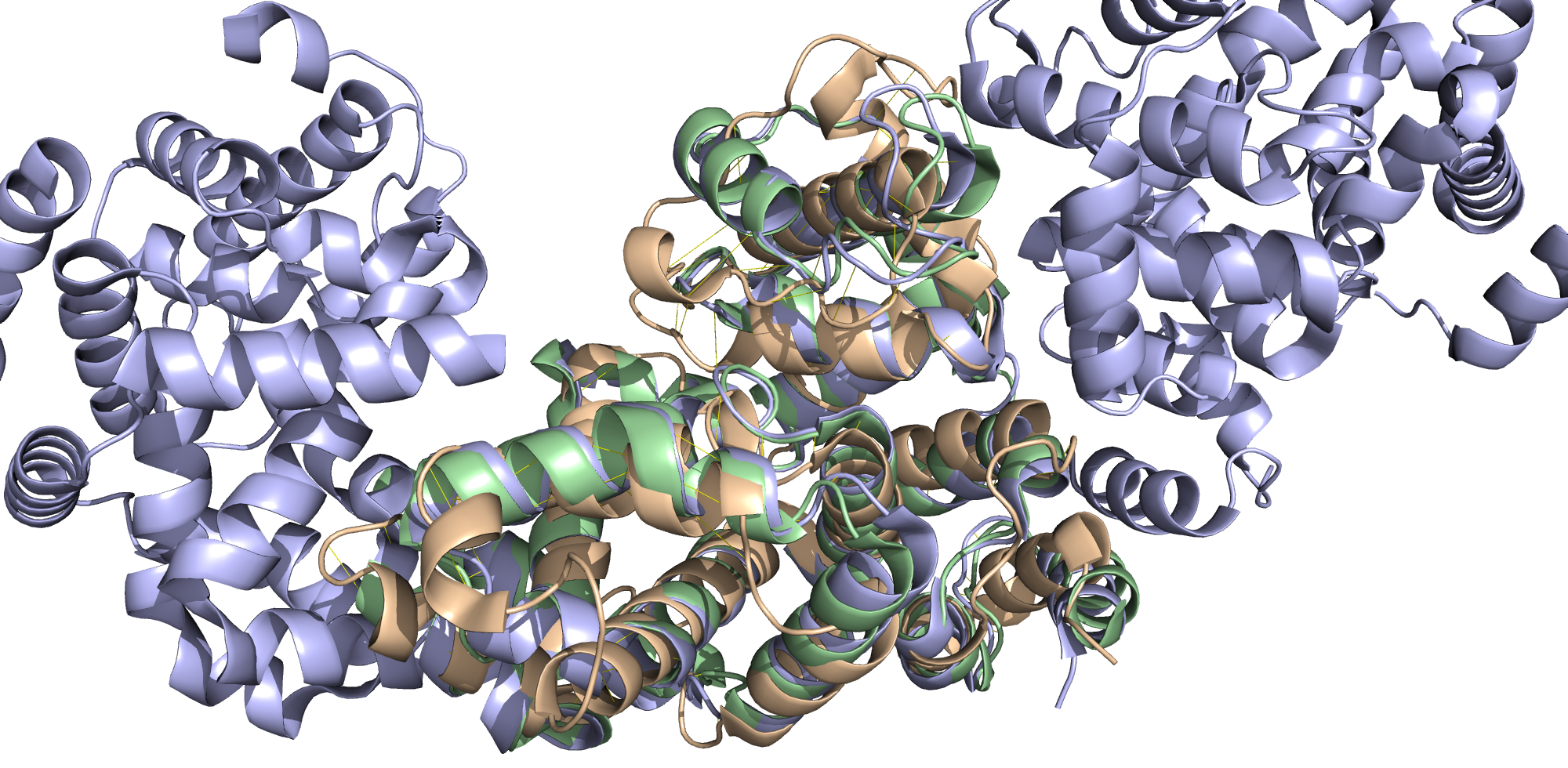
Как можно заметить, существует участок с высокой степенью гомологии структур (на нём сделан акцент на рис. 2). При этом мы наблюдаем и гетероморфность: так, например, у белка 3HZJ есть длинный хвост, который, вероятно, участвует в связывании. Такие рассуждения подтверждаются выравниванием программой ClustalW в JalView, которое можно найти тут. Также я сделала выравнивание на сайте PDB (вкладка Analyze; его можно найти тут), вывод представлен ниже. Всё это вновь подтверждает высокий уровень сходства последовательностей.

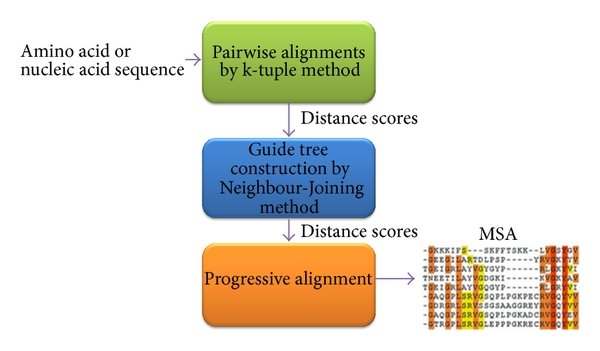
Clustal — это серия широко используемых компьютерных программ для множественного выравнивания последовательностей. У этого прогрессивного подхода есть две
основные проблемы:
– проблема локального минимума
– выбор параметров выравнивания.
ClustalW — это подход, призванный решить эти проблемы.
В ClustalW чувствительность
прогрессивного множественного выравнивания была значительно улучшена для выравнивания "расходящихся" белковых последовательностей. В новую программу включено несколько
модификаций. Эти модификации можно описать поэтапно:
[1] каждой последовательности при частичном выравнивании присваиваются индивидуальные веса, чтобы понизить вес [почти] повторяющихся последовательностей и
увеличить вес наиболее расходящихся. Эти весовые коэффициенты выполняются в соответствии с древовидной структурой MSA. Напротив, в обычном алгоритме
прогрессивного выравнивания, например, все последовательности будут иметь одинаковый вес.
[2] матрицы аминокислотных замен варьируются на разных стадиях выравнивания в соответствии с расхождением последовательностей, подлежащих выравниванию.
Чтобы вычислить оценку между положением из одной последовательности или выравнивания и положением из другой, используется среднее значение всех парных
оценок весовой матрицы для аминокислот в двух наборах последовательностей. Если какой-либо набор последовательностей содержит один или несколько пробелов
в одной из рассматриваемых позиций, каждый пробел по сравнению с остатком оценивается как ноль. Кроме того, используемые нами матрицы веса аминокислот по
умолчанию пересчитываются, чтобы иметь только положительные значения.
[3] штрафы за пробелы, специфичные для остатков, и локально уменьшенные штрафы за пробелы в гидрофильных областях стимулируют новые пробелы в областях потенциальных петель, а не регулярную вторичную структуру. Таким образом, эта обработка пропусков рассматривает оценку остатка по сравнению с пропуском как имеющую наихудшую возможную оценку. Наконец, позиции в ранних выравниваниях, где были открыты гэпы, получают локально уменьшенные штрафы за гэпы, чтобы стимулировать открытие новых гэпов на этих позициях.
[1] Higgins DG, Thompson JD, Gibson TJ. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996;266:383-402. doi: 10.1016/s0076-6879(96)66024-8. PMID: 8743695.
[2] Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002 Aug;Chapter 2:Unit 2.3. doi: 10.1002/0471250953.bi0203s00. PMID: 18792934.
[3] Che-Lun Hung, Yu-Shiang Lin, Chun-Yuan Lin, Yeh-Ching Chung, Yi-Fang Chung,
CUDA ClustalW: An efficient parallel algorithm for progressive multiple sequence alignment on Multi-GPUs.
[4] Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific
gap penalties and weight matrix choice. Nucleic Acids Res. 1994 Nov 11;22(22):4673-80. doi: 10.1093/nar/22.22.4673. PMID: 7984417; PMCID: PMC308517.