| Команда | Что делает |
|---|---|
| hisat2-build chr2.fasta | Индексация референса |
| fastqc chr2.fastq | Анализ программой FastQC.Получаем отчет в виде html файла |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr2.fastq chr2_trimmed.fastq TRAILING:20 MINLEN:50 | Очистка при помощи Trimmomatic. Чтения одноконцевые. Необходимо обрезать с конца нуклеотды с качеством ниже 20 и убрать все последовательности длиной менее 50. |
| hisat2 -x chr2_denwer02 -U chr8_trimmed.fastq -S chr8_aligntoref.sam --no-softclip --no-spliced-alignment | Картирование чтений из fastq файла на проиндексированную последовательность. --nosoftclip - выравнивание по всему риду; --no-spliced-alignment - выравнивание без гэпов и инделей в риде |
| samtools view -b chr8_aligntoref.sam -o chr8_aligntoref.bam | Перевод в bam формат |
| samtools sort chr8_aligntoref.bam chr8_altoref_sorted | Сортировка выравниваний по координате в референсе |
| samtools index chr8_altoref_sorted.bam | Индексирование |
| samtools mpileup -u -f chr8.fasta -o chr8_polymorph.bcf chr8_altoref_sorted.bam | Создание файла с полиморфизмами |
| bcftools call -cv -o chr2_polym.vcf chr2_polym.bcf | Изменение формата, -v - output variant sites only - показать только изменившиеся позиции |
| Координаты второго транскрипта | hg38 chr2:157,736,446-157,875,842 |
| vcftools --vcf chr2_polym.vcf --remove-indels --recode --out chr2_polym_noindel | Удалить из vcf файла индели |
| convert2annovar.pl -format vcf4 chr2_polym_noindel.recode.vcf -outfile chr2_polym.annovar | Конвертация в формат, который распознает annovar |
| annotate_variation.pl -outfile chr2_refgene -buildver hg19 -dbtype refGene chr2_polym.annovar | Аннотация по refgene, на генной разметке |
| annotate_variation.pl -filter -outfile chr2_dbsnp -buildver hg19 -dbtype snp138 chr2_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по dbsnp, основана на фильтрации |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -outfile chr2_1000g chr2_polym.annovar /nfs/srv/databases/annovar/humandb.old/ | Аннотация по 1000 genomes, основана на фильтрации |
| annotate_variation.pl -regionanno –buildver hg19 -outfile chr2_gwas -dbtype gwasCatalog chr2_polym.annovar /nfs/srv/databases/annovar/humandb.old/ | Аннотация по GWAS, основана на разметке других регионов генома |
| annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -outfile chr2_clinvar chr2_polym.annovar /nfs/srv/databases/annovar/humandb.old/ | Аннотация по Clinvar, основана на фильтрации |
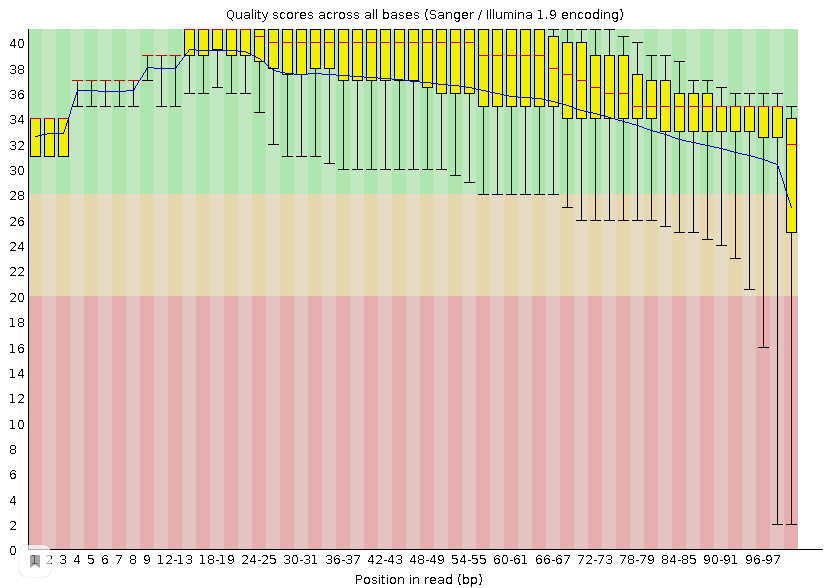
Рис. 1. Оценка качества чтений до триммирования
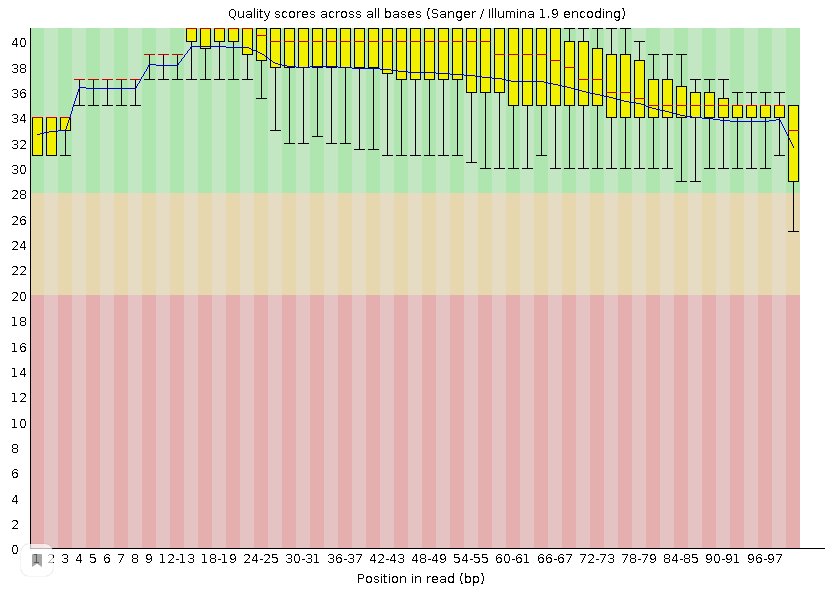
Рис. 2. Оценка качества чтений после триммирования
Было получено 10410 ридов длиной от 40 до 100 нуклеотидов, а среднее качество прочтения - около 38%. После очистки чтений их количество уменьшилось до 10191. На рисунках 1 и 2 можно видеть оценку качества чтений, полученную с помощью программы FastQC. По ним видно, что теперь качество каждого рида выше 20 и не располагается в красной области. Увеличилось среднее арифмитическое всех прочтений и общее качество данных немного улучшилось, поэтому триммирование оправдано. Результат работы Trimmomatic: TrimmomaticSE: Started with arguments: -phred33 chr2.fastq chr2_trimmed.fastq TRAILING:20 MINLEN:50 Input Reads: 10410 Surviving: 10191 (97,90%) Dropped: 219 (2,10%) TrimmomaticSE: Completed successfully Результат работы Hisat2: 10191 (100.00%) were unpaired; of these: 47 (0.46%) aligned 0 times 10140 (99.50%) aligned exactly 1 time 4 (0.04%) aligned >1 times 99.54% overall alignment rate 99,5% чтений выравнились 1 раз, всего 4 рида выравнились 2 и более раза и 47 прочтений, что составляет около 0,46% не выравнились. Можно сказать, что качество картирования довольно высокое.
Для описания snp и инделей был получен файл chr2_polym.vcf. Он содержит описания 79 полиморфизмов, из которых 7 - индели. Глубина покрытия указана в последнем столбце (DP), а качество покрытия можно посмотреть в столбце Qual. Все полученный данные занесены в таблицу 2. При анализе полиморфизмов были получены следующие значения: среднее арифмитическое - 70,46, медиана - 127,23, то есть ровно половина значений немного превышают медиану. Среднее значение глубины покрытий - 15,73, медиана - 3,5 (лучше характеризует этот показатель, так как примерно половина ридов накладываются на геном меньше четырех раз, но за счет участков с большой глубиной покрытия среднее арифмитическое завышено).
| Координата | Тип | Референс | Чтение | Глубина покрытия | Качество чтения |
|---|---|---|---|---|---|
| 234197717 | Замена | С | G | 26 | 104.008 |
| 234201186 | Замена | А | G | 14 | 126.008 |
| 234202274 | Вставка | ТСС | ТССС | 1 | 3.80767 |
Программа convert2annovar.pl для файла только с SNP показала, что из 72 замен 50 являются транзициями, а 22 трансверсиями. Далее производилась аннотация полученного файла по различным базам. При аннотации по refgene был получен файл chr2_refgene, в котором описано в каких участках ДНК произошли замены: exonic (6), UTR3 (6), UTR5 (2),ncRNA_exonic (1), intronic (57). Гены, в которые попали SNP: CCCDC88A, ATG16L1, MLPH. Для шести замен, попавших в экзоны в файле chr2_refgene.exonic_variant_function, cинонимичной является только попавшая в ген MLPH. Из аннотации по базе snp можно понять, сколько snp имееют rs. Информация лежит в файле snp138.hg19_snp138_dropped, получается, что 67 snp имеют rs, то есть это те замены, которые есть в банке snp. Оставшихся 5 замен в банке нет, они лежат в файле snp138.hg19_snp138_filtered. Также была посчитана средняя частота найденных snp по аннотации по базе 1000 Genomes. Она составила 37.7%, но она очень сильно варьируется, есть как часто встречающиеся snp, так и редкие. Одна аннотированная в Clinvar замена относится к заменам, вызывающим воспаление кишечника. В то же время, благодаря аннотации по GWAS, известно, что 2 полиморфизма ассоциированы с болезнью Крона, которое относится к воспалительным заболеваниям кишечника; однако оба snp находятся в гетерозиготном состоянии, поэтому нельзя точно говорить о фенотипическом проявлении этого варианта. Еще 1 полиморфизм вызывает рак простаты, и оставшийся полиморфизм скорее всего связан с особенностями роста (отмечен в gwasCatalog как "Height").