1. Описание файла с референсом
Это фаста файл с 7 хромосомой человека. Первая строка файла:>NC_000007.14 Homo sapiens chromosome 7, GRCh38.p13 Primary Assembly.NC_000007.14 - это идентификатор Refseq последовательности.
Об информиции в нём: в начале и конце последовательности имеется большое количество неопределённых нуклеотидов (N).
Исходя из того, что я знаю о строении хромосом, предположу, что это теломерные участки.
2. Индексация референса
С помощью команды:
bwa index -a bwtsw chr7.fna
Мы получаем 5 файлов: .amb .ann .bwt .pac .saС помощью мануала можно узнать, что параметр -a bwtsw позволяет выбрать алгоритм bwt-sw, позволяющий найти все локальные выравнивания. В chr7.fna.ann лежит краткая информация о последовательности.
В chr7.fna.amb - информация о нестандартных нуклеотидах (хотя вернее - о нуклеотидах, не являющихся "стандартными", т.е. G,T,A,C).
В chr7.fna.bwt - бинарный файл с последовательностью.
В chr7.fna.pac - сжатый файл.
В chr7.fna.sa - бинарный файл.
3. Описание образца
Я работал с файлами: SRR10720414_1.fastq.gz и SRR10720414_2.fastq.gz.Сслылка на информацию об образце в NCBI: ссылка
Прибор: Illumina Genome Analyzer IIx
Организм: Homo sapiens
Стратегия секвенирования: Whole-exome (экзомное)
Чтения: Парконцевые
Сколько чтений ожидается: 41,482,410
4. Проверка качества исходных чтений.
Использованная команда: fastqcКоличество пар чтений: 41482410
Совпадает ли количество чтений у “прямых” чтений и “обратных” чтений: Да.
Число пар чтений совпадает с ожидаемым
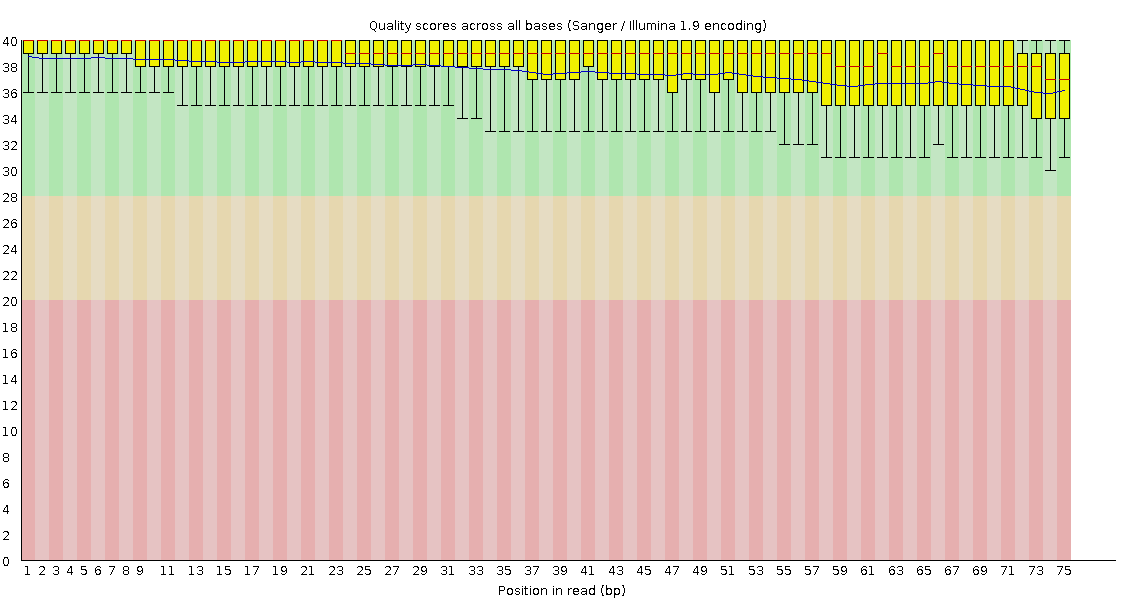
Картинки из раздела Per base sequence quality:
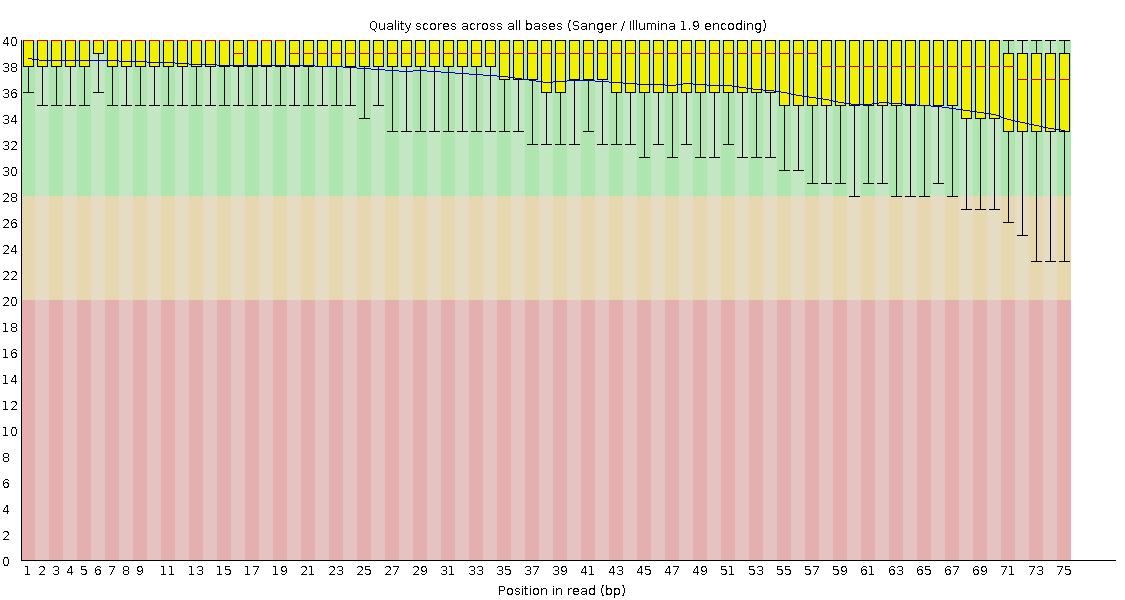
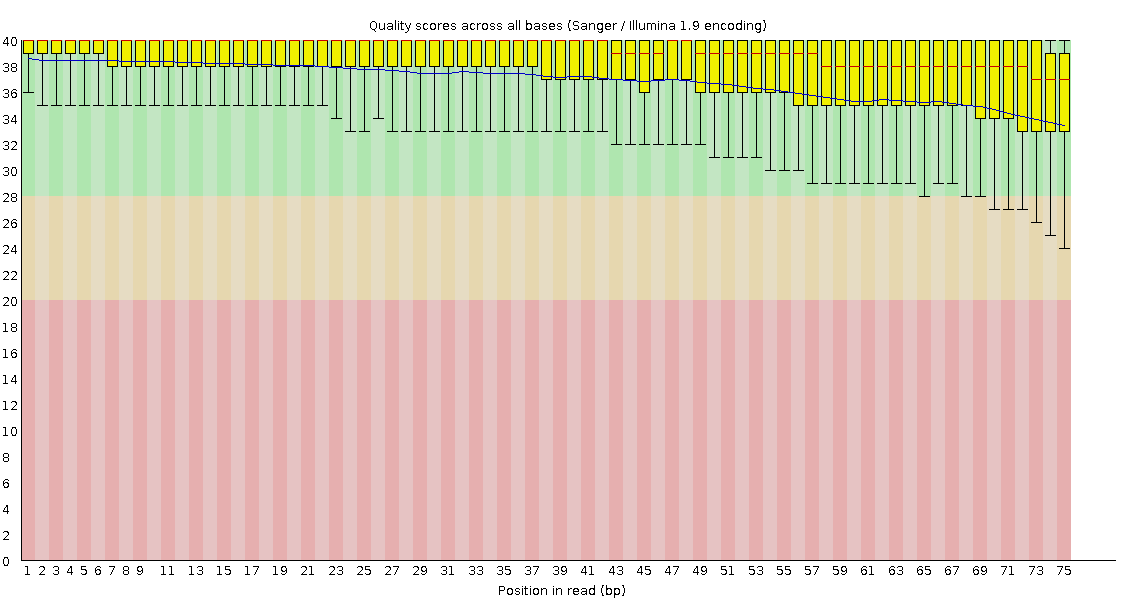
Картинки демонстрируют хорошее качество прочтения (значения находятся выше 30, что является хорошим результатом). Можно заметить, что качество чтения падает.
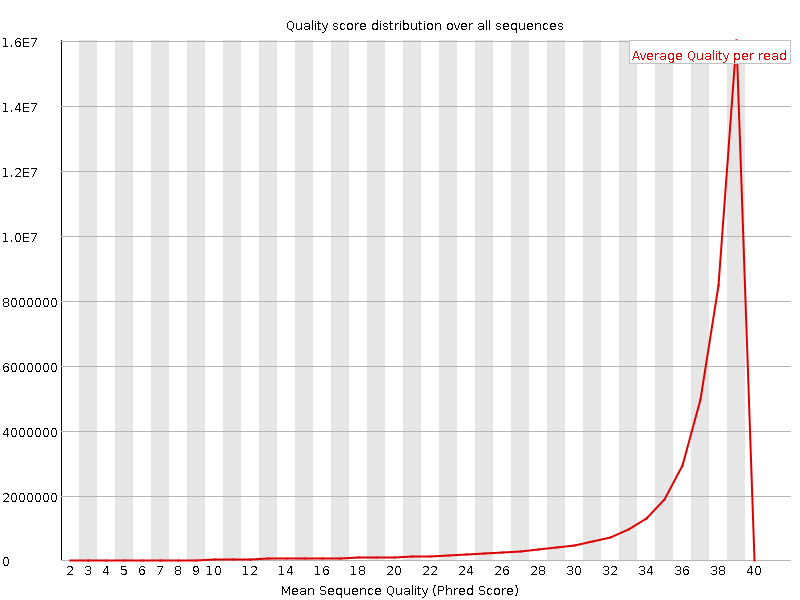
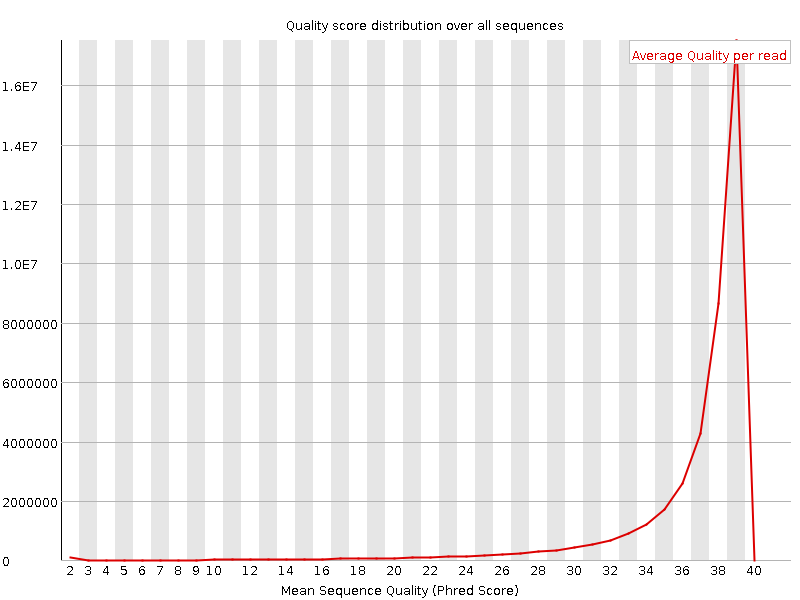
Данные графики показывают, что последовательности внутри основной нашей последовательности имеют высокое качество.
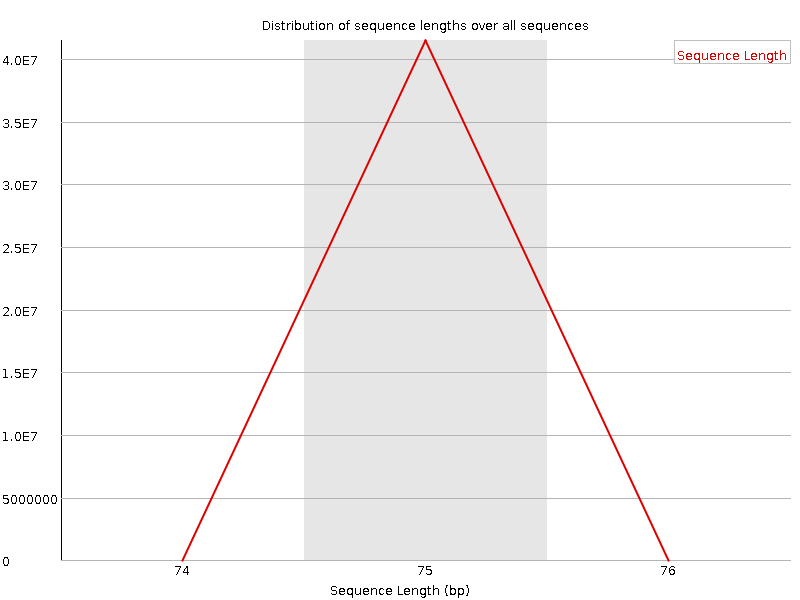
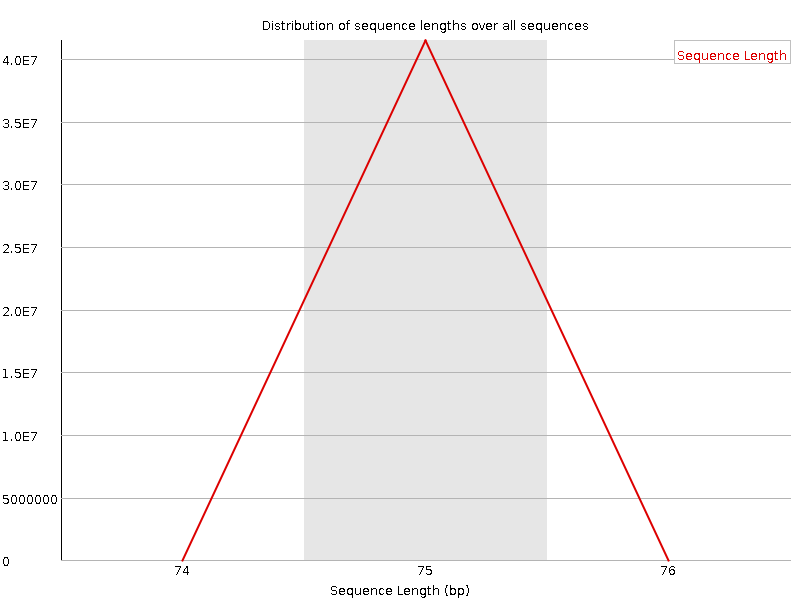
Судя по данным графикам - большинство чтений имело длину 74-76 нуклеотидов. Хотя если я правильно понял график, то тут все прочтения имели длину 75 нуклеотидов.
5. Фильтрация чтений.
Фильтрация с помощью trimmomatic, вот команда, поданная на вход:java -jar /usr/share/java/trimmomatic.jar PE -phred33 -threads 10 SRR10720414_1.fastq.gz SRR10720414_2.fastq.gz paired_1.fastq.gz unpaired_1.fastq.gz paired_2.fastq.gz unpaired_2.fastq.gz TRAILING:20 MINLEN:50
-threads 10 - Было задействованно 10 ядер.
TRAILING:20 - Удалить нуклеотиды, с качеством ниже, чем 20
MINLEN:50 - Удалить последовательности короче 50 нуклеотидов
6. Проверка качества триммированных чтений.
Использованная команда:fastqc paired_1.fastq.gz paired_2.fastq.gz unpaired_1.fastq.gz unpaired_2.fastq.gz
Вывод команды в командной строке:
Input Read Pairs: 41482410
Both Surviving: 39888569 (96.16%)
Forward Only Surviving: 674498 (1.63%)
Reverse Only Surviving: 704509 (1.70%)
Dropped: 214834 (0.52%)
TrimmomaticPE: Completed successfully
То есть количество оставшихся пар чтений: 39888569, что составляло 96.16% от начального.
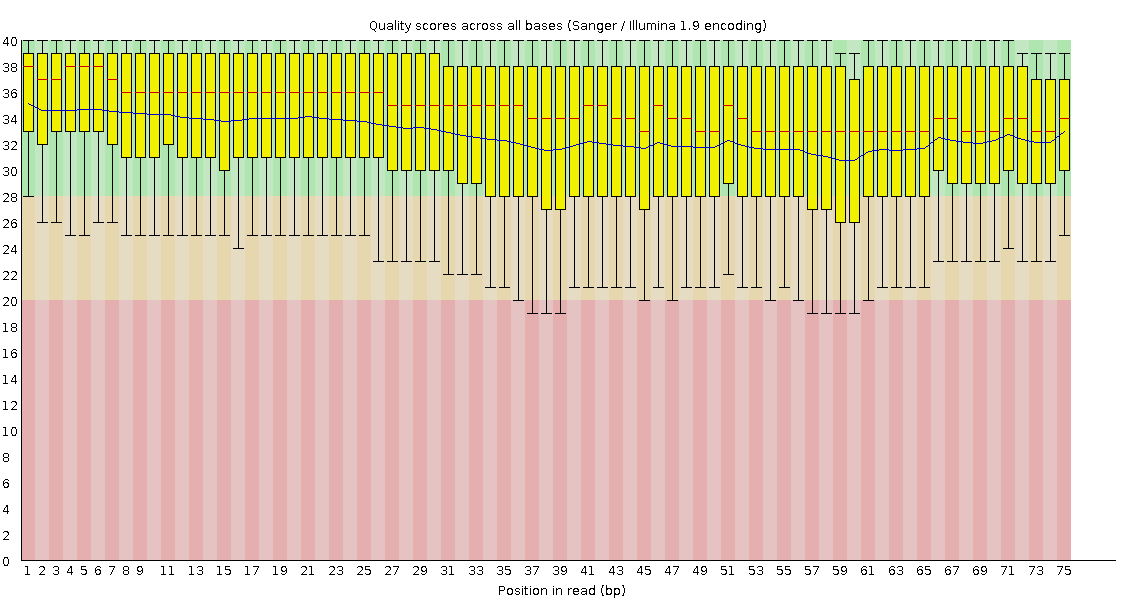
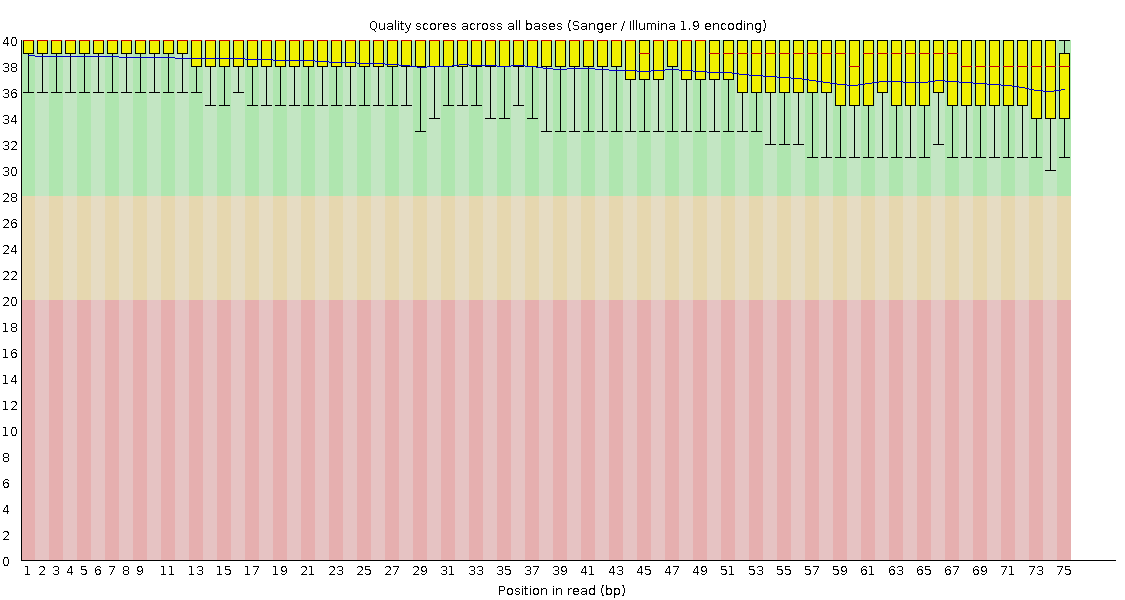
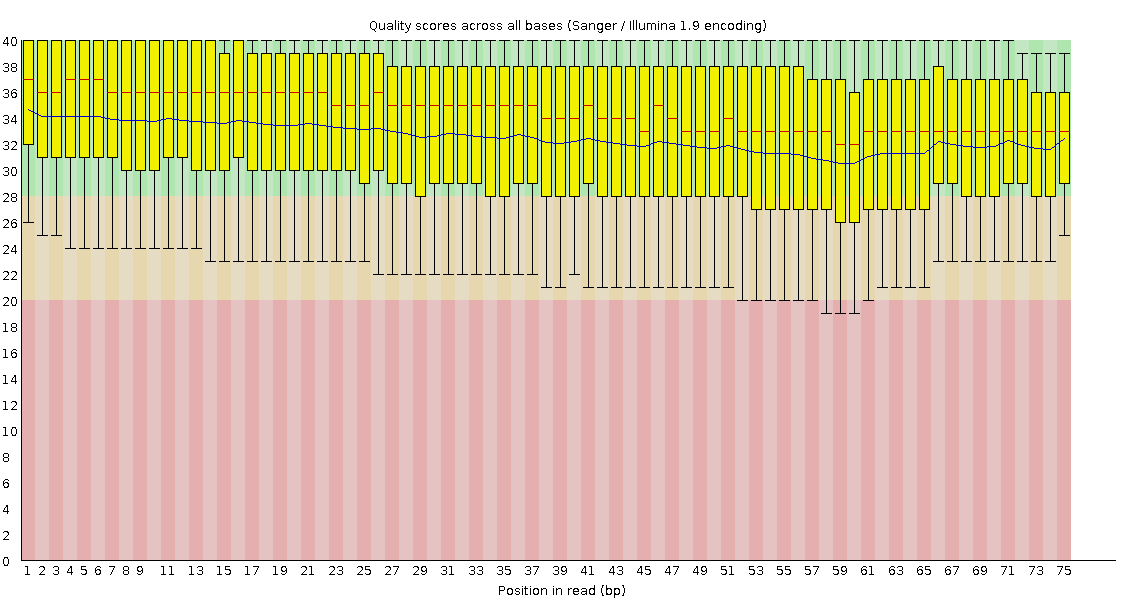
Заметно, что качество непарных заметно хуже по сравнению с парными.
Парные чтения стали заметно качественнее после фильтрации.
Картинки из раздела Per sequence quality scores (только для paired, 2 штуки):
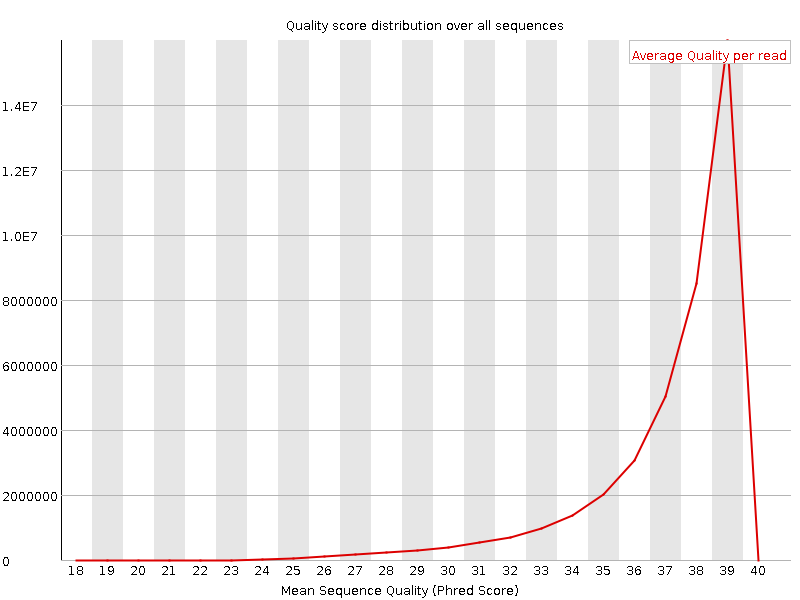
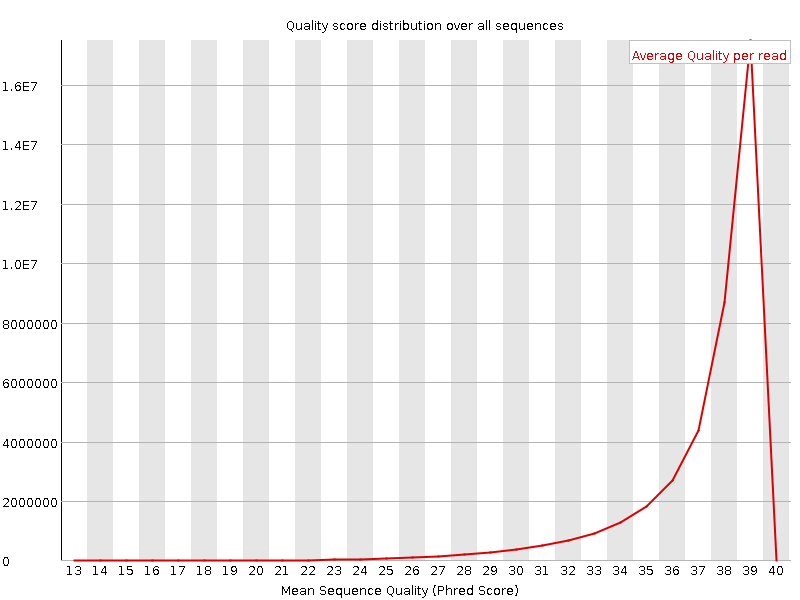
Начало графико стало короче, так как мы убрали короткие прочтения.
Картинки из раздела Sequence Length Distribution (только для paired, 2 штуки):
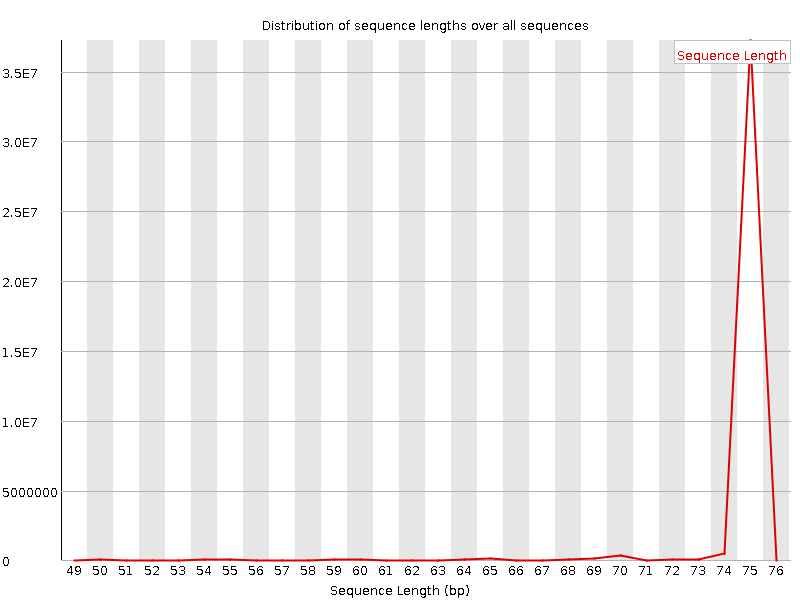
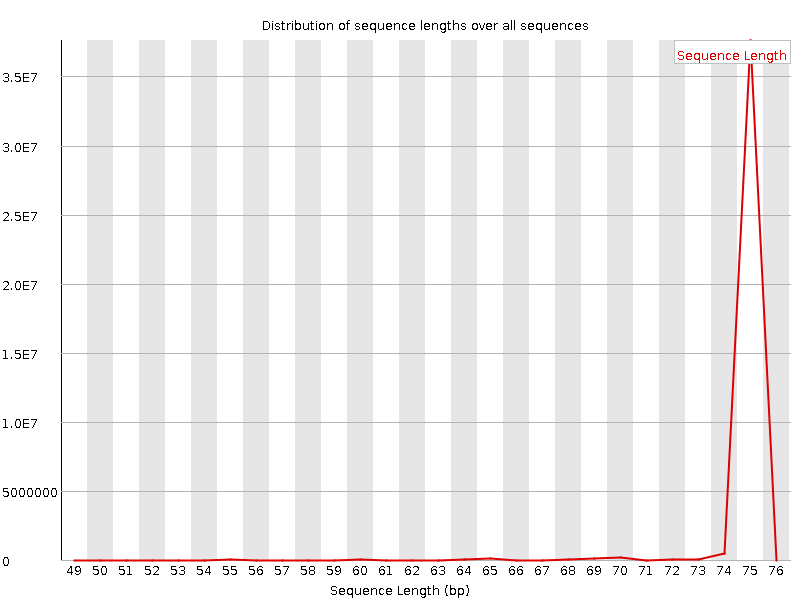
График стал заметно шире (появились более короткие чтения). Возможно это было связано с тем, что мы вырезали короткие прочтения.