EMBOSS
Last update on the 6th of November, 2017Several tasks in EMBOSS via CLI with few work under web-service and spreadsheet software. All the required files for each task are archived.
Task 1. Join fasta files.
File: task1.tar.gz
Input: mylist.txt
Output: joined.fasta
Line:
seqret -seq @mylist.txt -out joined.fasta
Task 2. Split fasta file.
File: task2.tar.gz
Input: coding1.fasta
Output: *.fasta
Line:
seqretsplit -seq coding1.fasta -auto
Task 3. Restrict 3 CDS from chromosome.
File: task3.tar.gz
Input: mylist.txt
Output: out.fasta
Line:
seqret @mylist.txt out.fasta
Task 4. Translate CDS.
File: task4.tar.gz
Input: in.fasta
Output: out.fasta
Line:
transeq -seq in.fasta -table 11 -out out.fasta
Task 5. Translate in six frames.
File: task5.tar.gz
Input: coding.fasta
Output: out.fasta
Line:
transeq -seq coding.fasta -frame 6 -table 11 -out out.fasta
Task 6. Transform alignment.
File: task6.tar.gz
Input: alignment.fasta
Output: align.msf
Line:
aligncopy -seq alignment.fasta -out align.msf -aformat2 msf
Task 7. Identical letters in alignment.
File: task7.tar.gz
Input: align.msf
Output: stdout
Line:
infoalign align.msf -only -refseq 2 -name -idcount -out stdout
Task 8. Transform feature table.
File: task8.tar.gz
Input: chromosome.gb
Output: table.gff
Line:
featcopy -fea chromosome.gb -outf table.gff
Task 9. Extract feature table.
File: task9.tar.gz
Input: seq.gb
Output: out.fasta
Line:
extractfeat -seq seq.gb -out out.fasta -type cds -describe product
Task 10. Shuffle sequence.
File: task10.tar.gz
Input: in.fasta
Output: out.fasta
Line:
shuffleseq in.fasta out.fasta
Task 11. BLAST of random sequence.
File: task11.tar.gz
Input: —
Output: —
Line:
makenucseq -amount 1 -out stdout | blastn -task blastn -db nt -outfmt 7 -out table.txt -remote
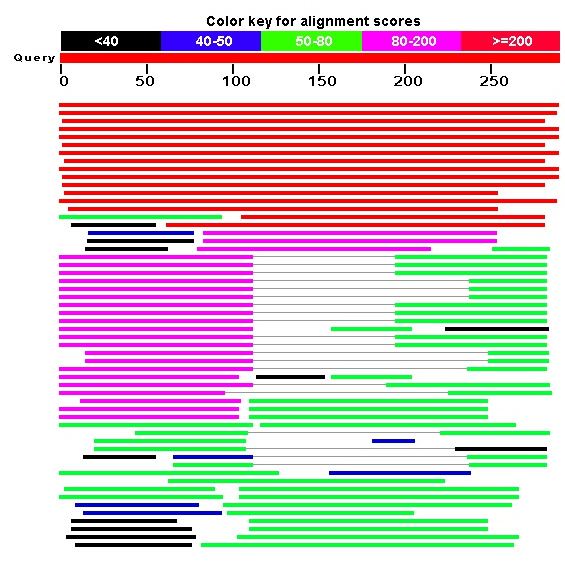
Blast+ on Kodomo is out-of-date and cannot connect to NCBI servers via https. So I copy-pasted sequence and ran BLAST via browser.
>EMBOSS_001 aacataaaggagcatgaaaaaacttttggaccagggaccctgtctcataacgctaacatc tagtgagctcgtctgtgtagcacatgcctagtgaagtgag
The result is shown below and in the hit_table.txt. Findings don't exhibit concordance concerning taxonomy and length of alignments isn't big that indicates probabilistic occurance of randomized sequence in nt bank. There were no "plausible" hits with E-value<0,1.
Task 12. Find ORFs and compare with "real".
File: task12.tar.gz
Input: seq.gb
Output: orf2.fasta
Line:
getorf -seq seq.gb -circular Y -reverse -methionine -minsize 300 -out orf2.fasta -find 3
Real CDS are written in real.fasta. With featcopy the feature table in gff format was extracted from
seq.gb. Information about predicted ORFs was extracted with infoseq. Then it was compiled
into table.ods and processed. Some stats are gathered in the table below.
| Property | Value |
|---|---|
| number_overlap | 2832 |
| overlap_forward | 1355 |
| overlap_reverse | 1477 |
| overlap/real | 0,646 |
| overlap/predicted | 0,473 |
| overlap_forward/real_forward | 0,632 |
| overlap_reverse/real_reverse | 0,659 |
| overlap_forward/predicted_forward | 0,454 |
| overlap_reverse/predicted_reverse | 0,493 |
As it is seen, more than half of "real" ORFs overlap with predicted. Share of matching ORFs in both forward and reverse strand is almost equal regarding real and predicted data. To assess density of matching ORFs the genome was binned into size of 10Kb and number of ORF medians was count for each bin. The result was plotted in the figure below.

It's clearly seen that almost all ORFs are distributed equally (between 4 and 8 peaks in bin) on the genome with few outliers.
Task 13. Codone frequencies.
File: task13.tar.gz
Input: gene_sequences.fasta
Output: out.txt
Line:
wordcount -seq gene_sequences.fasta -word 3 -out out.txt
Task 14. Dinucleotide frequencies in human chromosome.
File: task14.tar.gz
Input: chro.fa
Output: out.txt
Line:
compseq chro.fa -word 2 -out out.txt
9 dinucleotides were more frequent, than expected, the most frequent is AA.
Task 15. Align CDS regarding aligned proteins.
File: task15.tar.gz
Input: gene_sequences.fasta, protein_alignment.fasta
Output: out.fasta
Line:
tranalign -aseq gene_sequences.fasta -bseq protein_alignment.fasta -out out.fasta
Task 16. Local alignment of three sequences.
File: task16.tar.gz
Input: in.fasta
Output: align.fasta, out.edialign
Line:
edialign -seq in.fasta -outseq align.fasta -outfile out.edialign
Task 17. Remove gaps.
File: task17.tar.gz
Input: in.fasta
Output: out.fasta
Line:
degapseq -seq in.fasta -out out.fasta
Task 18. Carriage return.
File: task18.tar.gz
Input: set.txt
Output: out.txt
Line:
noreturn -in set.txt -out out.txt
Task 19. Random sequences.
File: task19.tar.gz
Input: —
Output: out.fasta
Line:
makenucseq -amount 3 -length 100 -out out.fasta -auto
Task 20. SRA to fasta.
File: task20.tar.gz
Input: sra_data.fastq
Output: data.fasta
Line:
seqret -seq sra_data.fastq -out data.fasta