В данном практикуме я также работал с хромосомой №10
Ниже представлена таблица со всеми использованными командами:
| Команда | Описание |
| fastqc chr10.1.fastq | Анализ качества чтений программой FastQC |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr10.1.fastq chr10.1_trimmed.fastq TRAILING:20 MINLEN:50 | Очистка при помощи Trimmomatic. Были обрезаны с конца нуклеотиды с качеством ниже 20, а также убраны все последовательности длиной менее 50 нк |
| hisat2 -x chr10_index -U chr10.1_trimmed.fastq -S chr10.1.sam --no-softclip | Картирование чтений на геном (--no-softclip – выравнивание должно быть по всему риду). Был убран параметр --no-spliced-alignment, так как он нужен для картирования без разрывов, а мы картируем последовательности рнк-транскриптов, а из них при созревании РНК вырезаются интроны, так что разрывы могут быть |
| samtools view -b chr10.1.sam -o chr10.1.bam | Перевод в .bam формат |
| sort chr10.1.bam chr10.1_sorted | Сортировка выравниваний по координате в референсе |
| samtools index chr10.1_sorted.bam | Индексирование |
| htseq-count -f bam -s no -i gene_id -m union chr10.1_sorted.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseq | Подсчёт чтений (-f - формат файла, -s - специфично ли исследование к считываемой цепочке (если нужно анализировать оба направления считывания, то no), -i - какие характеристики использовать для подсчета , -m - режим работы в неоднозначных ситуациях) |
| grep -wv 0 htseq > htseq.txt | Записывает в файл все строчки, значение для которых не 0 |
Триммирование
Исходно было 15462 чтений. После триммирования осталось 15313 (99.04%)Качество изначальных чтений изначально было весьма высоким, поэтому после обрезки качество сильно не изменилось. Было откинуто меньше 1,5% последовательностей. Поэтому, на мой взгляд, можно было обойтись и без триммировани.Качество до:
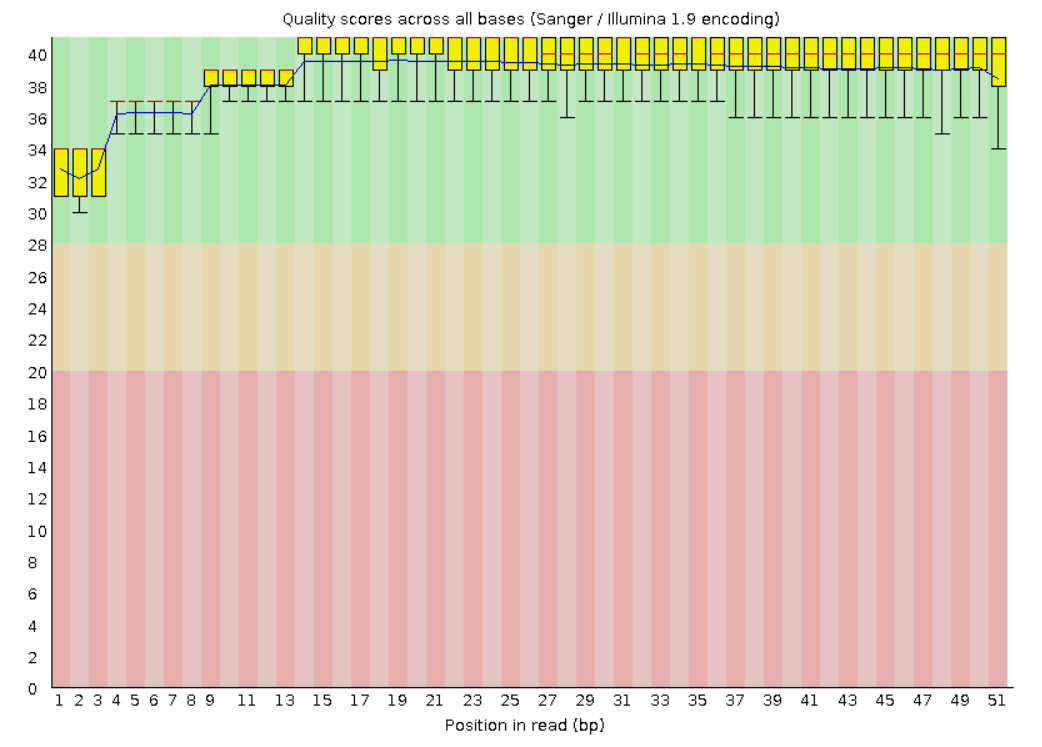
Качество после:
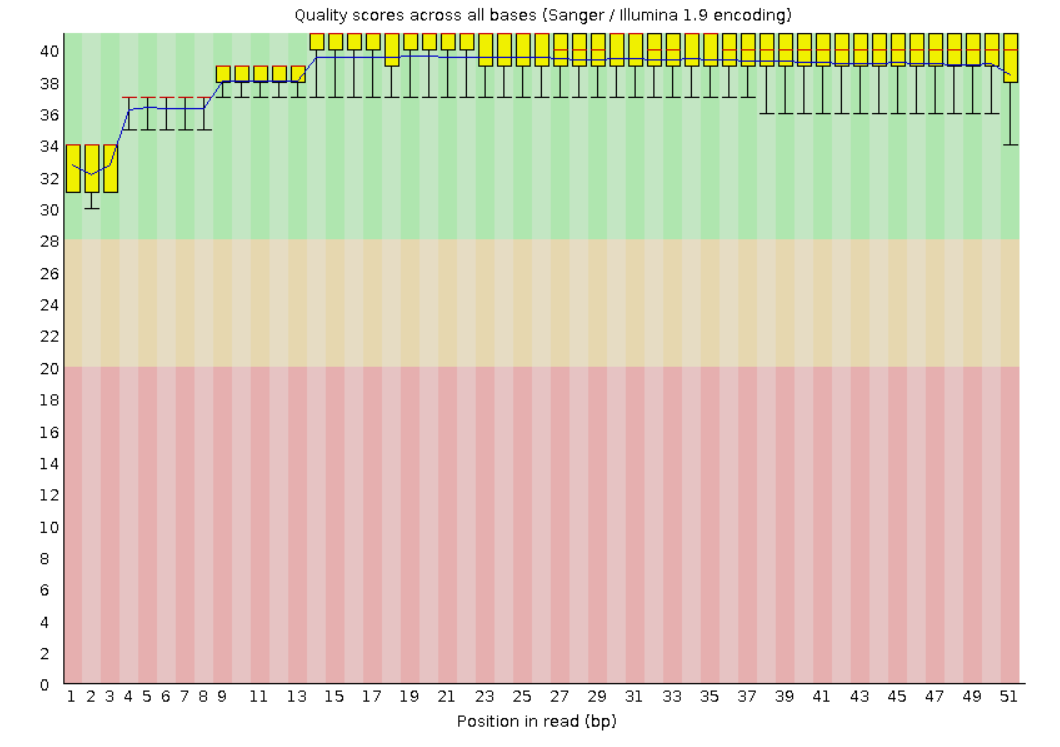
Картирование
Было откартировано 98.66% чтений. Результат работы программы hisat2:
15313 reads; of these:
15313 (100.00%) were unpaired; of these:
205 (1.34%) aligned 0 times
15108 (98.66%) aligned exactly 1 time
0 (0.00%) aligned >1 times
98.66% overall alignment rate
Можно сделать вывод, что это хорошее качество картирования
htseq-count
Результат работы программы:
ENSG00000165732.8 14501 ENSG00000266122.1 3 __no_feature 604 __not_aligned 205
Большинство прочтений попало в ген ENSG00000165732.8 (DDX21 | DExD-box helicase 21), который кодирует РНК-хеликазу, и всего 3 прочтения попало в ген ENSG00000266122.1, который является псевдогеном