Практикум №8
Задание 1. Использование разных вариантов BLAST для фрагмента ДНК.
Я решил взять для задания собаку (Canis lupus familiaris (dog)) и найти участок, куда попал бы ген инсулина (по нему точно гомологи найдутся). Идентификатор нуклеотидной записи: NC_049239.1 (18 хромосома).Координаты выбранного мной участка: 46778900..46780300 п.н. (длина 1400 нуклеотидов).
Ссылка на файл последовательности в формате fasta.
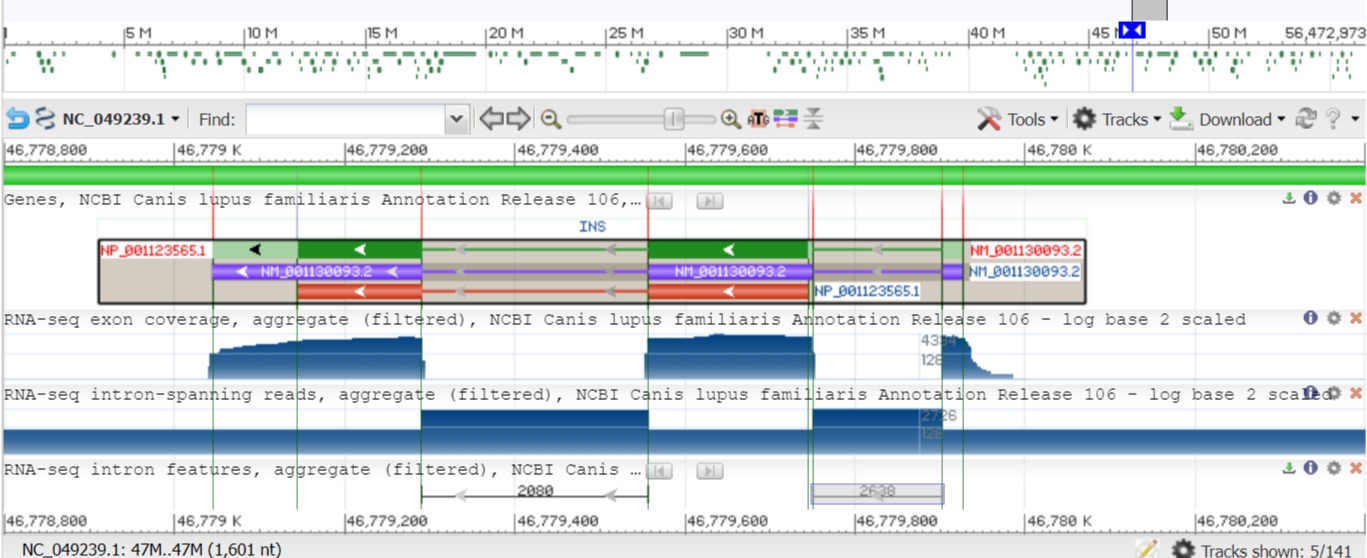
На участке есть ген инсулина ENSCAFG00845011141 (на комплементарной цепи). На рис. 1 ген показан зеленым, мРНК фиолетовым, участок, на котором располагается CDS - красным, длина самого CDS 333 нуклеотида (на хромосоме до сплайсинга занимает 600). Всего 2 интрона и 3 экзона.
Выбранный таксон - приматы (taxid:9443). Выбирал достаточно близкую группу, чтобы точно найти какие-то находки.
Blastn.
Программа выравнивает нуклеотидные последовательности. Параметры:
E-value: 0,05
Word size:11
Максимальное число находок:250
(оставил по умолчанию, так как качество и количество находок удовлетворило).
Получил 154 находки со средним процентом идентичности 70-80%, query cover сильно отличался, от 60% у находок
с E-value 7e-98 до 2% у находоки с E-value 0,025. В основном получившиеся находки это предсказанные последовательности, кодирующие белки (PREDICTED).
Megablast.
Программа также выравнивает нуклеотидные последовательности.
При этом megablast работает быстрее,
так как он ищет последовательности с высоким сходством.
Параметры:
E-value: 0,05
Word size:28
Максимальное число находок:250
Всего получил 108 находок, причем у всех средний query cover примерно 10-13%.
В основном предсказанные последовательности белков. Процент идентичности составляет в среднем 90%.
blastx.
Алгоритм отличается тем, что он заранее транслирует нуклеотидную последовательность в белковую,
и уже сравнивает эту белковую последовательность с белковыми базами данных. Это имеет смысл, так как
замены нуклеотидов могут быть синонимичными, в таком случае последовательность аминокислот
будет той же, хотя нуклеотидная изменилась. Параметры:
E-value: 0,05
Word size:5
Максимальное число находок:250
Матрица: BLOSUM62.
Всего нашлось 169 находок со средним процентом покрытия 15%, Значения E-value изменялись от
0.006 до 4e-19. Идентичность в среднем составила 90%. Как мы видим, количество находок больше,
отчасти это связано с вышеуказанной особенностью этой программы.
tblastx
Этот алгоритм сравнивает транслированную последовательность с другими транслированными послдеовательностями
ДНК. Это может быть полезно, так как для организма может не быть нужного белка в белковой базе данных.
E-value: 0,05
Word size:3
Максимальное число находок:100
Матрица: BLOSUM62.
К сожалению, blast не справился, а поставить более высокое значение word size нельзя...
Задание 2. Поиск генов основных рибосомальных РНК по далекому гомологу
Команда для индексации генома:
makeblastdb -in "C:\Dmitry\genome.fna" -dbtype nuclДля поиска гомологов я использовал алгоритм Blastn, потому что запросом и базой данных являются нуклеиновые кислоты, а tblastx и blatsx не подходят, потому что транслирование рРНК не имеет биологического смысла, megablast не использовал, т.к. последовательности не являются родственными. Использовал параметры -evalue для того, чтобы отобрать наиболее значимые находки, word_size (для обоих взял умолчательные значение из ncbi blast), также использовал -outfmt=7 для того, чтобы получить результаты выравниваний в табличном виде.
Локальный blast запустил с помощью команд:
blastn -task blastn -query "C:\Dmitry\16s.fasta" -evalue 0.05 -word_size 11 -db "C:\Dmitry\genome.fna" -out 16s.out -outfmt=7
blastn -task blastn -query "C:\Dmitry\23s.fasta" -evalue 0.05 -word_size 11 -db "C:\Dmitry\genome.fna" -out 23s.out -outfmt=7Ссылка на файл выдачи с 16s
Ссылка на файл выдачи c 23s
В ncbi полное название rRNA-16S ribosomal RNA и rRNA-23S ribosomal RNA. Это РНК, которые входят в состав рибосом (16s в малую, 23s в большую) цифра и буква s означает константу седиментации. 3′-конец 16S РНК содержит последовательность анти-Шайна-Дальгарно, с помощью которой 16S рРНК связывается с мРНК; 23s РНК содержит пептидил-трансферазный центр.
Полученные результаты:
Анализ выдачи с 16SСразу бросается в глаза то, что характеристика выравниваний отличается только координатами последовательности из базы данных + если не использовать опцию -outfmt=7 можно увидеть, что выравнивания абсолютно идентичны. Первая находка с AC NW_023331920.1 и координатами 23220..23262, как оказалось, находится внутри гена, кодирующего рРНК 18S, у самого гена координаты 21397..23265 (нашел в ncbi nucleotides). Далее в аннотации сборки Refseq нашел те же самые данные. Сама находка это нелокализованный скаффолд. Во второй находке - NW_023331571.1 я получил те же самые данные. Но продукт этих генов был предсказан компьютером (Derived by automated computational analysis using gene prediction method) Из последней находки NC_049228.1 в ncbi nucleotides находим, что данная последовательность находится в 7 хромосоме, внутри гена, кодирующего белок CCDC178 (coiled-coil domain containing 178). С чем связано такое несоответствие, я затрудняюсь ответить, даже если бы гены располагались на разных цепях это было бы указано на графике в ncbi nucleotides. Но интуитивно кажется, что в данном случае последовательности действительно гомологичны.
Анализ выдачи с 23SЗдесь тоже есть странности...Рассмотрю случай двух идентичных выравниваний
NC_049253.1 (40221707..40221536) оказался в 17 хромосоме
Gene: LOC119877817
Qualifiers: Pseudo, Partial start
Location: complement(40,221,031..40,225,481)
Length: 4,451 nt
NC_049238.1 (65082001..65081830) тоже в 17 хромосоме
Gene: LOC119877202
rRNA: XR_005372833.1
Length: 4,740 nt
Name: rRNA-28S ribosomal RNA