Поиск в геноме участков, кодирующих белки, похожие на заданный
Создала в своей рабочей директории индексные файлы пакета BLAST+ для поиска по заданному геному бактерии Streptococcus agalactiae. Выбрала подходящую для решения данной задачи программу из пакета BLAST+ (tblastn) и провела с ее помощью поиск с порогом на E-value 0,001. По результатам поиска заполнила таблицу 1.
Таблица 1. Поиск гомологов белка YWHB_BACSU в геноме S.agalactiae.
| Число находок с E-value < 0,001 | 1 |
| E-value лучшей находки | 2e-08 |
| Название последовательности с лучшей находкой | embl|AL766848|AL766848 |
| Координаты лучшей находки (от-до) | 205302-205466 |
| Доля последовательности моего белка, вошедшая в выравнивание с лучшей находкой | 41.82 |
Поиск гомологов некодирующих последовательностей программой BLASTN
Запустила программу blastn, указав в качестве последовательностей для поиска файл trna_bacsu.fasta, в качестве банка – геном бактерии, отформатированный в предыдущем задании. Установила табличный формат выдачи (опция "-outfmt 7"). Порог на E-value установила равным 0,01. Просмотрела выходной файл. Придумала, как запустить grep так, чтобы на выходе получилось число – количество находок именно для данной последовательности grep "\# [0-9]*". Создала колонку из названий входных последовательностей командой grep ">" trna_bacsu.fasta. Импортировала её в Excel.
Поиск гомологов при изменённых параметрах программы BLASTN
Повторила предыдущее задание ещё два раза с изменёнными параметрами программы, каждый раз сохраняя результаты в новый файл. В первый раз изменила весовую матрицу, то есть параметры -reward и -penalty. Установила -reward 5 и -penalty 4. При этом поменяла также параметры -gapopen и -gapextend на -gapopen 8 и -gapextend 6. Во второй раз, оставив те же значения параметров -reward, -penalty, -gapopen и -gapextend, поменяла также значение параметра -word_size на минимально возможное - 4.
Как дополнительное задание сделала ещё один столбец с результатами поиска при минимальном значении word_size и взятыми по умолчанию параметрами вычисления веса выравнивания.
Результатом этого и предыдущего упражнений стал Excel-файл trna.xls, лежащий в директории H:\term3\block3\BLAST. Также данный файл можно скачать по ссылке.
Использованные команды:
time blastn -task blastn -query trna_bacsu.fasta -db sa_genome.fasta -evalue 0.01 -word_size 11 -outfmt 7 > 1111 real 0m0.570s user 0m0.364s sys 0m0.200s time blastn -task blastn -query trna_bacsu.fasta -db sa_genome.fasta -evalue 0.01 -word_size 11 -outfmt 7 -reward 5 -penalty -4 -gapopen 8 -gapextend 6 > 1112 real 0m0.679s user 0m0.420s sys 0m0.256s time blastn -task blastn -query trna_bacsu.fasta -db sa_genome.fasta -evalue 0.01 -word_size 4 -outfmt 7 -reward 5 -penalty -4 -gapopen 8 -gapextend 6 > 1113 real 0m21.151s user 0m20.900s sys 0m0.224s time blastn -task blastn -query trna_bacsu.fasta -db sa_genome.fasta -evalue 0.01 -word_size 4 -outfmt 7 > 1114 real 0m15.953s user 0m15.700s sys 0m0.244s
Анализ результатов
На рисунке 1 показана зависимость числа найденных гомологов от изменения параметров расчёта веса выравнивания и изменения длины слова.
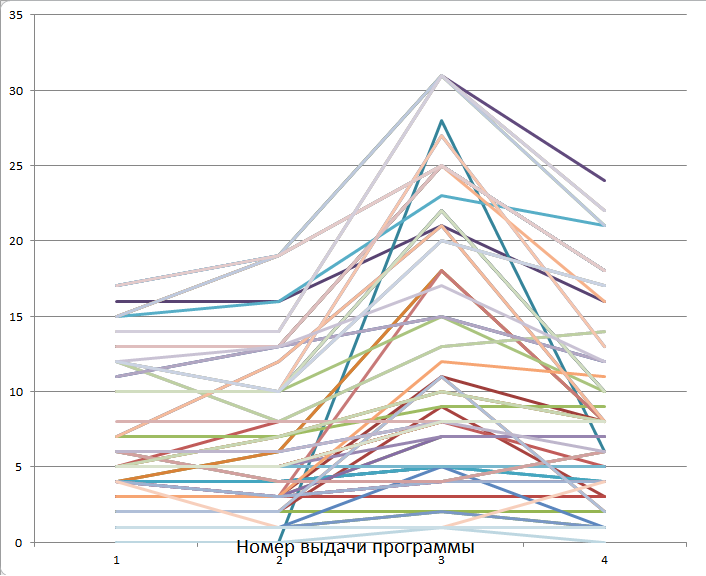
Рисунок 1. Распределение числа найденных гомологов при изменении параметров расчёта веса выравнивания и изменении длины слова. По вертикальной оси находится количество находок, по горизонтальной оси - номер выдачи программы, согласно файлу trna.xslx.
На данном графике можно проследить тенденцию к небольшому увеличению числа гомологов при изменении параметров -reward 5 и -penalty 4. Можно отметить резкое увеличение числа найденных последовательностей при уменьшении длины слова и небольшое снижение числа гомологов при восстановлении первоначальных параметров веса матрицы. Тем самым наиболее "лояльным" можно признать поиск с параметрами -word_size 4 -outfmt 7 -reward 5 -penalty -4 -gapopen 8 -gapextend 6
Выбрала пару tRNA-Thr B.subtilis и гомологичный участок, найденный в геноме Streptococcus agalactiae. Выбрала находку, которая находится программой BLASTN при самых мягких параметрах и не находится при других. Это связано с тем, что последовательность этой тРНК не выравнивается словом длиной 7 - есть участки, отличающиеся друг от друга незначительно, но они меньшей длины. Вырезала гомологичный участок в отдельный файл командой seqret -sask.Привожу в отчете выравнивание программой needle.
Aligned_sequences: 2
# 1: AL766845
# 2: BSn5_t20996
# Matrix: EDNAFULL
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 73
# Identity: 58/73 (79.5%)
# Similarity: 58/73 (79.5%)
# Gaps: 0/73 ( 0.0%)
# Score: 230.0
#
#
#=======================================
AL766845 1 gccggcttagctcagttggtagagcatctgatttgtaatcagagggtcgc 50
|||||..|||||||.||||||||||..||||.||||||.|||.||.|.|.
BSn5_t20996 1 gccggtgtagctcaattggtagagcgcctgacttgtaagcagtggattgg 50
AL766845 51 gtgttcaagtcatgtagccggca 73
|.|||||||||.|.|.|||||||
BSn5_t20996 51 gggttcaagtcctcttgccggca 73
#---------------------------------------
#---------------------------------------
Из выравнивания можно понять, что последовательности схожи. Их процент идентичности довольно высок, гэпы отсутствуют. Имеются незначительные различия в нескольких нуклеотидах. Так как структура тРНК консервативна, а выравнивание имеет большой вес, то можно предположить, что выявленные различия не мешают выполнять тРНК свои функции.
Гомологичный участок в поле FT записи EMBL, описывающей геном бактерии проаннотирован как тРНК, следовательно, программа работает хорошо.
FT tRNA 88985..89057 FT /product="transfert RNA-Thr"
Время работы программы BLAST
Проследила за изменением времени работы программы при изменении параметров, для этого при запуске на kodomo в командной строке перед всей командой написала слово time. Время работы программы от заданных параметров зависит примерно следующим образом: меньше секунды тратится на выполнение команд со стандартными параметрами или с измененной весовой матрицей. Наиболее сильно приводит к увеличению времени выполнения команды (вплоть до 30 раз) укорачивание длины слова при поиске, что оправдывается более точными результатами.