Алгоритмы и программы множественного выравнивания
СРАВНЕНИЕ ВЫРАВНИВАНИЯ ОДНИХ И ТЕХ ЖЕ ПОСЛЕДОВАТЕЛЬНОСТЕЙ ТРЕМЯ РАЗНЫМИ ПРОГРАММАМИ
Построение множественного выравнивания выполнялось для белковых доменов (seed) семейства PF00007 из практикума 11. Последовательности выравнивались с помощью сервисов JalView, выбранные программы - MUSCLE, MAFFT и TCOFFEE (с параметрами по умолчанию), сравнивались MUSCLE и TCOFFEE, MUSCLE и MAFFT (референсным выравниванием было выбрано выполненное программой MUSCLE, наиболее часто используемой в учебных отчетах программой). Поскольку сервис сравнения множественных выравниваний VerAlign на момент выполнения отчета не работал по неизвестным причинам, полученные выравнивания сравнивались с помощью программы, написанной моей однокурсницей Гончаровой Еленой.
| Участок | MUSCLE и MAFFT | MUSCLE и TCOFFEE |
|---|---|---|
| Совпадение №1 | (14,69)=(14,69) | (72,90)=(75,93) |
| Несовпадение №1 | (1,13)=(1,13) | (1,18)=(1,18) |
| Совпадение №2 | (72,90)=(72,90) | (19,26)=(19,26) |
| Несовпадение №2 | (70,71)=(70,71) | (27,31)=(27,31) |
| Совпадение №3 | (93,96)=(93,96) | (32,36)=(32,36) |
| Несовпадение №3 | (91,92)=(91,92) | (37,43)=(37,43) |
| Совпадение №4 | - | (44,45)=(44,45) |
| Несовпадение №4 | - | (48,71)=(46,47) |
| Совпадение №5 | - | (46,47)=(48,49) |
| Несовпадение №5 | - | (91,94)=(50,74) |
| Совпадение №6 | - | (95,96)=(98,99) |
| Несовпадение №6 | - | (97,113)=(94,97) |
| Cовпадение №7 | - | - |
| Несовпадение №7 | - | (-,-)=(100,120) |
| Одиночные совпадающие колонки вне блоков | - | (39,39)=(39,39) |
Гиперссылка на файл с проектом Jalview.
TCOFFEE-fasta, MAFFT-fasta, MUSCLE-fasta: гиперссылки на fasta-файлы с множественными выравниваниями, выполненными с помощью различных программ.
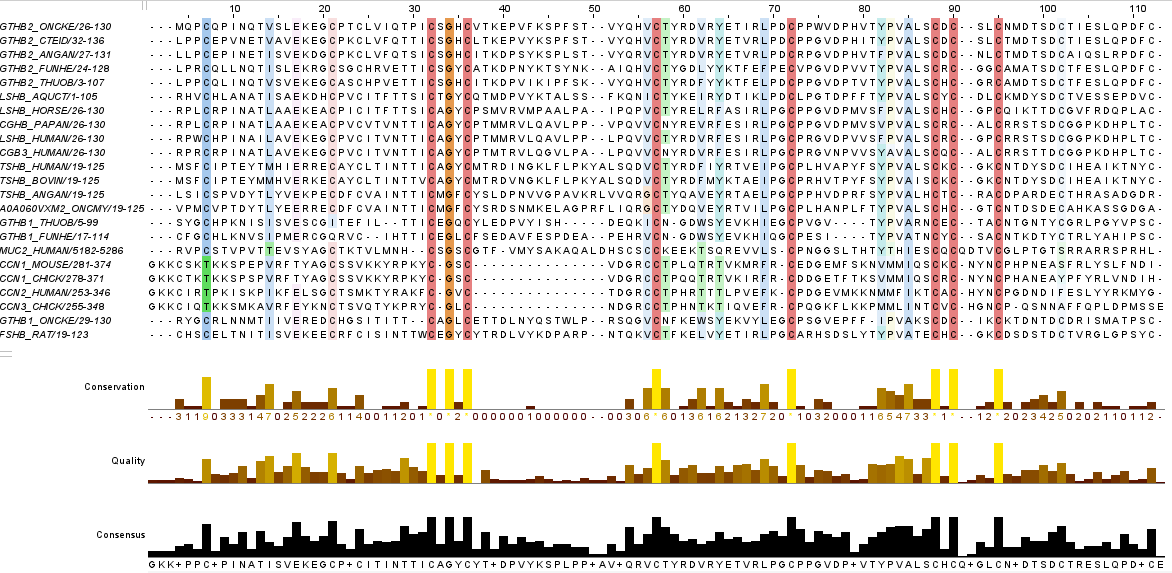
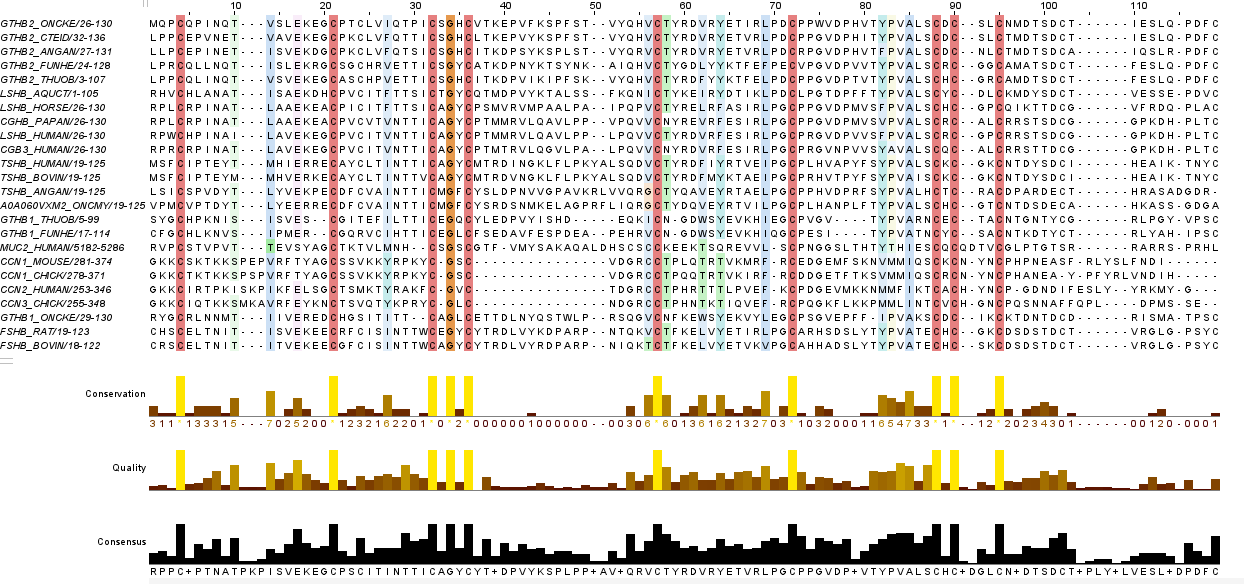
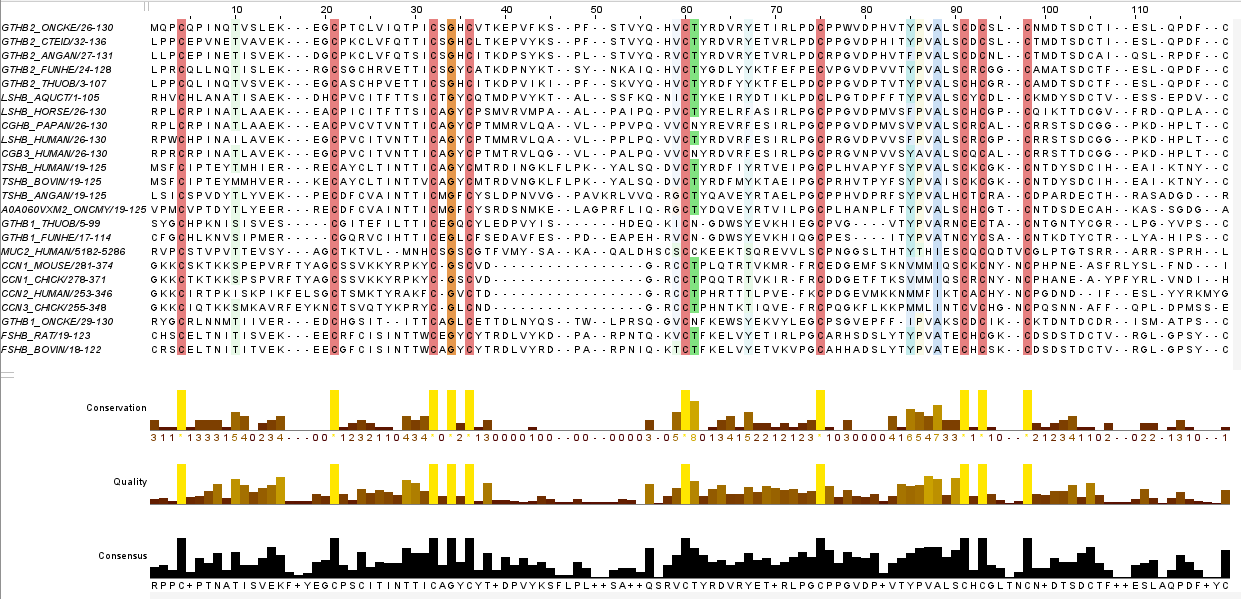
Основываясь на визуальном анализе выравниваний и информации, полученной с помощью программного анализа участков совпадений и несовпадений, можно заключить, что выравнивания, сделанные программами MUSCLE и TCOFFEE, имеют больше совпадающих блоков, нежели выравнивания, сделанные программами MUSCLE и MAFFT. Несмотря на это, согласно результату работы скрипта, процент и абсолютное число совпадающих колонок для MUSCLE и TCOFFEE ниже, чем для MUSCLE и MAFFT (39 колонок и 34,5% и 32,5% от длин выравниваний для MUSCLE и TCOFFEE и 79 колонок и 66,4% и 69,9% от длин выравниваний для MAFFT и MUSCLE). Получается, что MUSCLE и TCOFFEE похожи большим числом некрупных блоков, тогда как MUSCLE и MAFFT имеют крупные общие блоки в небольшом количестве. Если же основываться в целом на проценте схожих колонок, то алгоритмы MUSCLE и MAFFT можно считать более близкими по смыслу, чем MUSCLE и TCOFFEE. Несмотря на различия, все выравнивания выявляют схожие высококонсервативные участки, что указывает на идейное сходство их алгоритма.
ПОСТРОЕНИЕ ВЫРАВНИВАНИЯ ПО СОВМЕЩЕНИЮ СТРУКТУР И СРАВНЕНИЕ ЕГО С ВЫРАВНИВАНИЕМ MSA
Для выполнения задания было выбрано три белка из PF00007: 1hrp, 5bpu, 8enf.
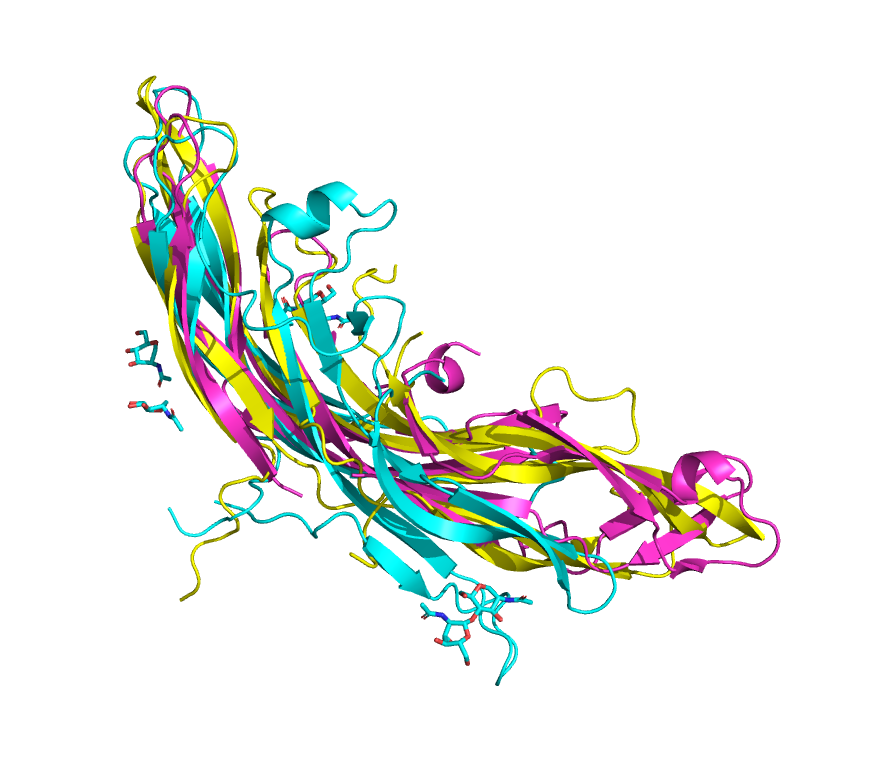
С помощью программы PDBeFold было получено множественное совмещение структур (см. рис. 4). Полученное выравнивание в формате fasta было перевыровнено программой MUSCLE согласно комментариям к практикуму 12. Два полученных выравнивания анализировались аналогично с помощью программы Елены Гончаровой.
| Участок | PDBeFOLD и MUSCLE |
|---|---|
| Совпадение №1 | (1,19)=(1,19) |
| Несовпадение №1 | (20,23)=(20,23) |
| Совпадение №2 | (68,82)=(66,80) |
| Несовпадение №2 | (28,35)=(28,35) |
| Совпадение №3 | (91,105)=(89,103) |
| Несовпадение №3 | (41,67)=(41,65) |
| Совпадение №4 | (117,124)=(114,121) |
| Несовпадение №4 | (83,84)=(81,82) |
| Совпадение №5 | (36,40)=(36,40) |
| Несовпадение №5 | (87,90)=(85,88) |
| Совпадение №6 | (24,27)=(24,27) |
| Несовпадение №6 | (106,116)=(104,113) |
| Совпадение №7 | (85,86)=(83,84) |
| Одиночно совпадающие колонки вне блоков | (22,22)=(22,22) |
И структурное наложение, и MUSCLE-выравнивание указывают на высокую схожесть выбранных белков, особенно основных мотивов бета-тяжевой структуры. Небольшие различия наблюдаются в положении неупорядоченных участков, а также в положении небольших альфа-спиральных мотивов у 1hrp и 8enf (которые у 5bpu вообще отсутствуют), и в меньшем размере 1hrp, из-за чего у него отсутствует небольшой участок консервативной у 8enf и 5bpu бета-листовой структуры.
Основываясь на визуальном анализе выравниваний и длинах совпадающих и несовпадающих участков, их количестве, а также на проценте совпадающих колонок от длины выравниваний (55,6% и 57,0% по результатам работы скрипта) можно заключить, что программы выравниваний достаточно похожи. Оба алгоритма выделяют практически идентичные консервативные участки близкого размера, выравнивания отличаются в большинстве своем лишь расположением гэпов (в ситуациях, где оно вариативно). Однако, судя по выравниванию С-концов белков, PDBeFold имеет тенденцию к вставлению большего числа гэпов и открытию большего числа инделей, что делает его выравнивание несколько длиннее выравнивания MUSCLE.
Гиперссылка на файл с проектом Jalview.
PDBeFold-fasta, MUSCLE-fasta: гиперссылки на fasta-файлы с множественными выравниваниями, выполненными с помощью различных программ.
КРАТКОЕ ОПИСАНИЕ ОДНОЙ ИЗ ПРОГРАММ MSA
Для выполнения задания была выбрана программа MUSCLE как наиболее часто используемая в практических работах этого семестра.
MUSCLE (multiple sequence comparison by log‐expectation) — компьютерная программа для множественного выравнивания белковых последовательностей [1], алгоритм которой был впервые описан в 2004 году Робертом К. Эдгаром.
Алгоритм MUSCLE близок к алгоритму работы MAFFT: он так же требует построения направляющего дерева, после чего происходит попарное выравнивание профилей, которое используется сначала для прогрессивного выравнивания, а затем для уточнения. Таким образом, алгоритм включает в себя три этапа: предварительное прогрессивное выравнивание, которое создается быстро, но с относительно низкой точностью (для последовательностей попарно вычисляются расстояния, определяемые числом совпадающих k-меров в них, которые собираются в матрицу расстояний, кластеризуемую по методу UPGMA с получением бинарного дерева), улучшенное прогрессивное выравнивание, при котором дерево перестраивается с поправкой на вычисление расстояния между последовательностями более точным способом (т.н. "расстояние Кимуры"), уточнение итогового выравнивания с выбором результатов с наивысшей оценкой.
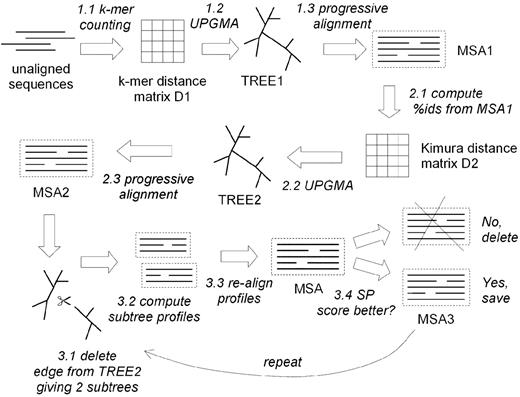
Согласно статистическим тестам, основанным на проверках, проведенных автором алгоритма, MUSCLE по точности на 1-2.5% выше алгоритма TCOFFEE, однако работает быстрее при анализе большого числа последовательностей, превышая по скорости также алгоритм CLUSTALW [2]. Несмотря на высокую эффективность работы, алгоритм MUSCLE имеет ограничения, так как он не учитывает, какие именно изменения аминокислот происходят в последовательностях [3]. Это обстоятельство может быть и плюсом при построении выравниваний близких последовательностей, поскольку подобная программа, предполагающая одинаковое наказание за любую замену аминокислоты на ранних этапах прогрессивного выравнивания, позволяет избежать увеличения расстояния между парами. Еще один недостаток MUSCLE заключается в снижении точности выравнивания при работе с консерватиными блоками, которые встречаются в небольшом числе входных последовательностей [4]. Тем не менее, относительно разумный баланс между скоростью работы и точностью выравнивания делает MUSCLE широко используемым алгоритмом множественного выравнивания.
ЛИТЕРАТУРА
[1] Robert C. Edgar, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Research, Volume 32, Issue 5, 1 March 2004, Pages 1792–1797, https://doi.org/10.1093/nar/gkh340
[2] R. C. Edgar, "MUSCLE: multiple sequence alignment with improved accuracy and speed," Proceedings. 2004 IEEE Computational Systems Bioinformatics Conference, 2004. CSB 2004., Stanford, CA, USA, 2004, pp. 728-729, doi: 10.1109/CSB.2004.1332560.
[3] Pais, F.SM., Ruy, P.d.C., Oliveira, G. et al. Assessing the efficiency of multiple sequence alignment programs. Algorithms Mol Biol 9, 4 (2014). https://doi.org/10.1186/1748-7188-9-4
[4] A Comprehensive Benchmark Study of Multiple Sequence Alignment Methods: Current Challenges and Future Perspectives. Julie D. Thompson, Benjamin Linard, Odile Lecompte, Olivier Poch. Published: March 31, 2011 https://doi.org/10.1371/journal.pone.0018093