Практикум 12
Сравнение выравниваний
Для выполнения последнего практикума я выбрал белки из 9 практикума, т.к уже выравнивал их:PURU_ECOLI, PURU_BACSU, PURU_MYCBO, PURU_SHIFL, PURU_CORS1, PURU_HAEIN.
Для сравнения я выбрал 3 программы - ClustalWS, Muscle и Mafft.
За референс я взял программу ClustalWS (почему-бы нет?).
Ссылки на выравнивания:
Работал с программой Ксении Кирцовой, за что ей большое спасибо. Результаты следующие:
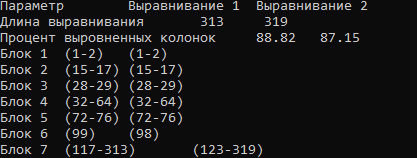
Рис.1 Это результат сравнения Clustal и Mafft.
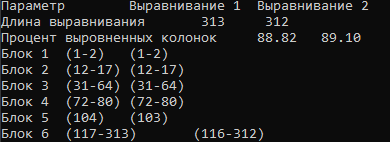
Рис.2 А это сравнение Clustal с Muscle
Как можно заметить, результаты получились довольно схожие. Длина выравнивания одинакова у первого выравнивания (313), но отличается у второго (319-Clustal и Mafft, 312-у Clustal и Muscle)
Пространственное выравнивание и его сравнение с MSA выравниванием
Для выполнения этого задания я брал 3D структуры из семейства доменов, которое я выбрал в 11 практикуме: BCCT (betaine/carnitine/choline family transporter)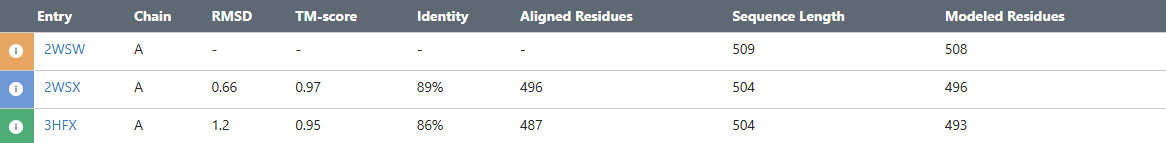
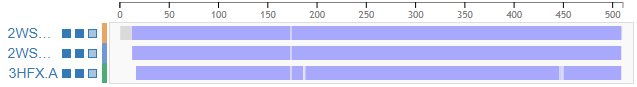
Рис.3-4 Результаты пространственного выравнивания белков 2WSW, 2WSX и 3HFX.
Ссылки на:
Для итогового сравнения я еще раз воспользовался программой Ксении, получив следующие результаты:
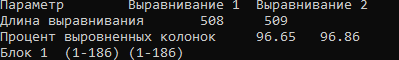
Рис.5 Сравнение пространственного выраванивания и выравнивания с помощью ClustalWS.
В итоге результаты получились очень схожими, длина отличалась совсем незначительно, как и процент выровненных колонок.
ClustalWS
ClustalWS (Clustal W and Clustal Omega) — это семейство программ, предназначенных для многократного выравнивания последовательностей (Multiple Sequence Alignment, MSA).
Программа ClustalWS используется для выравнивания белков, РНК и ДНК-последовательностей, что позволяет выявить эволюционные связи между ними.
Основные этапы включают:
1. Вычисление матрицы расстояний: Вычисляется сходство между каждой парой последовательностей, обычно с помощью таких методов, как метод Нидлмана-Вунша или метод Смита-Ватермана.
2. Создание филогенетического дерева: На основе матрицы расстояний строится дерево, представляющее эволюционные связи между последовательностями.
3. Пошаговое выравнивание: Последовательности выравниваются в соответствии с деревом, начиная с наиболее близких пар и постепенно добавляя более далекие последовательности.
ClustalWS позволяет исследователям анализировать последовательности на генетическом уровне, выявляя общие мотивы, функциональные области и эволюционные изменения. Программа широко используется в молекулярной биологии, геномике и биоинформатике.