В таблице 1 приведены основные данные белков, для которых выполнялись задания.
| Entry | Entry name | Protein names | Length | Organism | Superkingdom |
| Q07US6 | DNAK_RHOP5 | Chaperone protein DnaK (HSP70) | 633 | Rhodopseudomonas palustris (strain BisA53) | Bacteria |
| A1T2S3 | DNAK_MYCVP | Chaperone protein DnaK (HSP70) | 622 | Mycobacterium vanbaalenii (strain DSM 7251 / PYR-1) | Bacteria |
| O65719 | HSP7C_ARATH | Heat shock 70 kDa protein 3 | 649 | Arabidopsis thaliana (Mouse-ear cress) | Eukaryota |
| P27541 | HSP70_BRUMA | Heat shock 70 kDa protein | 644 | Brugia malayi (Filarial nematode worm) | Eukaryota |
| Q9HRY2 | DNAK_HALSA | Chaperone protein DnaK (HSP70) | 629 | Halobacterium salinarum (strain ATCC 700922 / JCM 11081 / NRC-1) (Halobacterium halobium) | Archaea |
| Q97BG8 | DNAK_THEVO | Chaperone protein DnaK (HSP70) | 613 | Thermoplasma volcanium (strain ATCC 51530 / DSM 4299 / JCM 9571 / NBRC 15438 / GSS1) | Archaea |
Таблица 1.Основные данные.
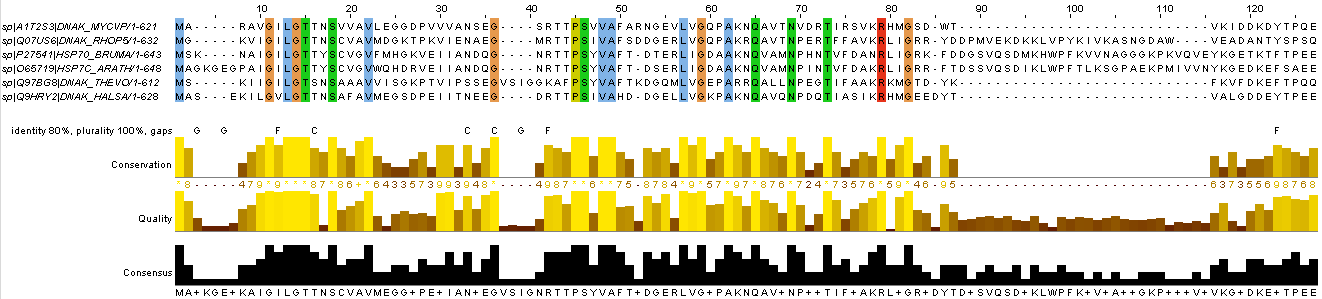
Ниже приведено выравнивание (программа JalView, Tcoffee with Defaults) последовательностей белков с раскраской по схеме ClustalX с условием Identity Threshold = 100%. На рисунке 1 приведена часть выравнивания (для просмотра его целиком щелкните на картинку или скачайте файл). Помимо выравнивания была добавлена новая строка разметки (identity 80%, plurality 100%, gaps), в которой было отмечено по три примера разных типов консервативности: консервативных на 80% или более (C), абсолютно функционально консервативных (F), позиций с гэпами (G).
Рисунок 1. Выравнивание и разметка.
С помощью команды infoalign пакета EMBOSS были посчитаны следующие параметры выравнивания: Name-SeqLen (имя и длина последовательности), AlignLen (длина выравнивания), GapLen (число гэпов), % (процент гэпов от длины выравнивания), Ident (число идентичных позиций), Similar (число похожих на идентичные позиций), % (процент суммы идентичных и похожих на них позиций от длины выравнивания). Данные были получены для консервативности и функциональной консервативности 100% (таблицы 2 и 3) и более 70% (таблицы 4 и 5). Функционально консервативной считается позиция, в которой стоят только аминокислотные остатки со схожими свойствами (схожими функциональными группами). Например, ароматические аминокислоты (триптофан, тирозин, фенилаланин) или аминокислоты с аминогруппой в раликале (аргинин, лизин). Удивителельно, что при функциональной консервативности 100% у последовательностей отличается число идентичных позиций. Однако так выдает программа. Но если прибавить к этим значениям значения колонки Similar, то процент везде будет одинаков. Видимо, так и надо сделать.
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Таблица 2.Параметры при консервативности 100%. | Таблица 3.Параметры при функциональной консервативности 100%. |
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Таблица 4.Параметры при консервативности 70%. | Таблица 5.Параметры при функциональной консервативности 70%. |
В таблице 5 представлены сводные данные (для значений при консервативности 70% взяты средние значения параметров с уже прибавленными значениями столбца Similar).
| AlignLen | GapLen | % | Identity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| identity 100% | % | plurality 100% | % | identity 70% | % | plurality 70% | % | |||
| 692 | 60,33 | 8,72 | 169 | 24,42 | 279 | 40,32 | 311,18 | 45,06 | 357,33 | 51,64 |
Таблица 6. Сводные данные по выравниванию.
Ссылки: весь проект (выравнивание из задания 1, автоматическое и исправленное выравнивания из задания 2) можно скачать по ссылке.
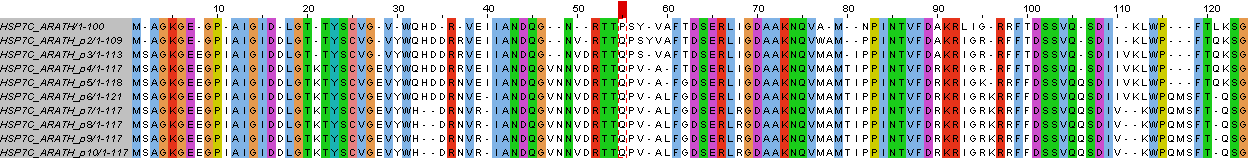
Рисунок 2. Автоматическое выравнивание.
Однако, полученное выравнивание не всегда соответствует эволюции (самому вероятному ее варианту). Ниже приведен список мест, которые скорее всего ей не соответствуют.
Исправление мест, где выравнивание не соответствует эволюции:
И само исправленное выравнивание можно увидеть на рисунке 3 (на сей раз выравнивание раскрашено по схеме ClustalX без порога)
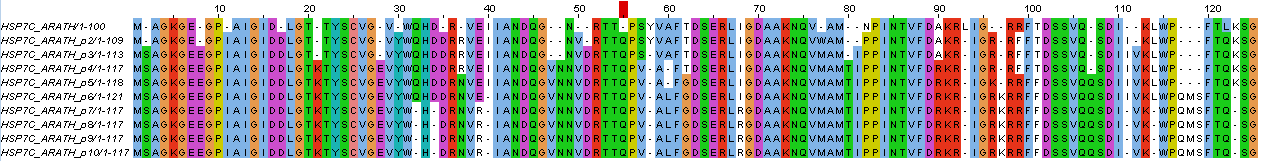
Рисунок 3. Исправленное выравнивание.
Первые 10 мутаций:
Для одного из белков (а именно A1T2S3 - шаперона из Mycobacterium vanbaalenii) была найдена его нуклеотидная последовательность (на сайте NCBI был найден геном бактерии, а из нее вырезан соответствующий фрагмент последовательности - ID белка: WP_011777910.1). Далее так же, как и в предыдущем задании были проведены 10 раундов мутаций (только в этот раз везде сновилось по 6 штук). Полученные последовательности были транскрибированы с помощью команды transeq покета EMBOSS и объединены в 1 файл (см. скрипт). Далее последовательности были импортированы в Jalview, выровнены (с помощью Tcoffee with Defaults) и выравнивание раскрашено по схеме ClustalX с условием Identity Threshold = 50%. Ниже представлен фрагмент выравнивания, чтобы увидеть его целиком - щелкните на изображение.
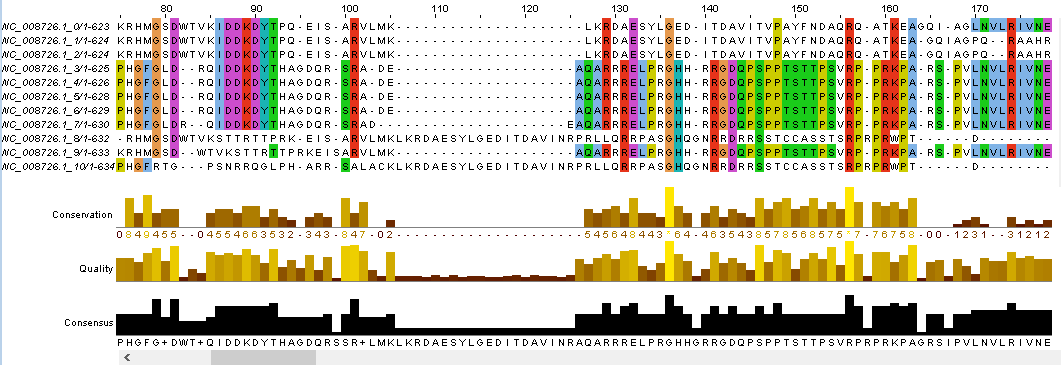
Рисунок 4. Выравнивание нуклеотидных последовательностей.
На приведенном фрагменте видно, что выравнивание здесь работает куда хуже, чем в случае с аминокислотными последовательностями (чтобы удостовериться в этом, можете посмотреть на таблицы 7 и 8). На нем также видно, что последовательности разбиваются на группы, местами абсолютно совпадающие без всяких гэпов (как показано на рисунке 5). Сильные различия (куда сильнее, чем в случае с аминокислотными последовательностями) объясняются, скорее всего, тем, что происходит сдвиг рамки считывания (из-за делеции нуклеотида или, наоборот, его вставки). В этом случае понятно, что не имеет смысла выравнивание по местами случайно совпадающих аминокислот, и, следовательно, добавление кучи гэпов. Гигантские изменения могла вызвать всего 1 мутация, что означает, что крайне низкие значения консервативности тоже мало что значат (и точно не значат, что это кардинально разные последовательности). Я исправила выравнивание (точнее заново выровняла - потому что поменялся принцип выравнивания, теперь цель - совместить одинаковые фрагменты последовательностей, игнорируя то, что между ними не совпадает), ниже приведен фрагмент этого выравнивания - тот же, что и на рисунке 4 (чтобы посмотреть полное выравнивание щелкните на рисунок 5 или скачайте файл).
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Таблица 7.Параметры при консервативности 70%. | Таблица 8.Параметры при функциональной консервативности 70%. |

Рисунок 5. Исправленное выравнивание.
Первые 10 мутаций в этих последовательностях (с позиции 13 и далее, начиная с p3) как раз объясняются сдвигом рамки считывания (то есть на самом деле только одной мутацией): к изначальной последовательности AAC UCA... - которая соответствует аминокислотам N и S, добавился нуклеотид G, и получившаяся последовательность GAA CUC A... - как раз соответствует аминокислотам E, L, R...
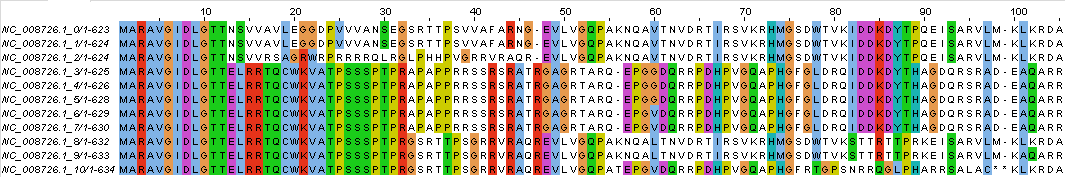
Рисунок 6. Начало исправленного выравнивания - первые мутации.
1) "Последовательность белка обычно под стабилизирующем отбором, т.е. отбор действует против мутаций а.к.о"
В книге А. Маркова "Рождение сложности" (Глава 7. Управляемые мутации, раздел "Частота мутаций под контролем") описывается SOS-response бактерий, при котором они намеренно увеличивают частоту появления мутаций. Делают они это в экстренных условиях для повышения разнообразия и, следовательно, вероятности, что как-то удачный мутант выживет. В числе включающихся SOS-генов есть ген dinB, который кодирует склонную к ошибкам ДНК-полимеразу, которая и отвечает за увеличение частоты мутирования. В той же книге в следующем разделе - "Целенаправленное создание новых генов" - рассматривается еще более известный процесс - внесение мутаций в гены антител в В-лимфацитах (или соматическое гипермутирование). В обоих случаях клетки находятся под давлением отбора (немутировавшие бактерии умирают, неподходящие лимфоциты уничтожаются), который никак не действует против мутаций.
2) "Мутации происходят постоянно и случайно"
Контрпримером может служить все то же соматическое гипермутирование. Все в той же книге описан его механизм:"Он [этот процесс] идет под контролем специальных ферментов и имеет отчасти "закономерный", а отчасти "случайный" характер. Ключевую роль играет особый фермент — цитидин-дезаминаза, который атакует нуклеотиды Ц (цитозины) в V-области гена антитела и превращает их в урацилы (У). Как мы помним, урацил в норме входит в состав РНК, но не ДНК. Присутствие урацила в цепи ДНК служит "сигналом тревоги" для ферментов, осуществляющих репарацию — починку поврежденных участков ДНК. Эти ферменты находят урацил и начинают "исправлять ошибку". При этом они вырезают вокруг урацила довольно большой кусок ДНК (длиной до 60 нуклеотидов), а потом восстанавливают его, но делают это очень неаккуратно. В результате такой неточной починки вокруг того места, где находился урацил, возникает множество мутаций." То есть с одной стороны, мутации в итоге вносятся случайно, с другой стороны, их появление не случайно, они появляются только в определенных местах, и изначально изменяются только цитозины. Все это не случайно.
3)"Только мутации в половых клетках наследуются"
В этом предложении содержится два утверждения, к одному из которых точно можно привести контрпример - в половых клетках наследуются не только мутации. В "Рождении сложности" в главе 7 ("Наследуются ли приобретенные признаки?") разобраны несколько вариантов эпигенетического наследования - наследования признаков без мутирования генетического материала. Это и метилирование дочерней ДНК, и состав материнских матричных РНК в яйцеклетке (а потом и в зиготе), и другие механизмы.
Ко второму утверждению - мутации только в половых клетках наследуются - также можно найти контрпример в "Рождении сложности", в главе 3 ("Великий симбиоз"). В разделе "Лучшее — детям: как клопы кормят свое потомство ценными симбионтами" разбирается случай насекомых (тлей и щитников) и их симбионтов (бактерий), эволюционировавших параллельно.
У обоих групп насекомых симбионты передаются по наследству - от матери к потомству, то есть новое поколение наследует симбионтов с их изменениями генетического материала. И наследование это очень строгое - личинки без симбионтов гораздо менее жизнеспособны и между особями симбионты не передаются. А абсолютная синхронность эволюции хозяев и симбионтов и вовсе заставляет задать вопрос: изменение насекомого заставляло менться бактерию, или наоборот, изменения в бактерии меняли ее хозяина? Но всяко связь между ними очень крепка. Так что можно это рассматривать как раз как наследование мутаций не в половых клетках.
4) "В гомологичных последовательностях живущих сегодня организмов мы видим почти исключительно мутации, прошедшие отбор"
За исключением уже рассмотренных выше случаев, тут стоит упомянуть следующее. Безусловно, при наличии отбора, сохранятся будут только те мутации, которые этот отбор поддержит. Однако отбор, скорее всего, действует не на весь геном, ведь гены - на которые как раз действует отбор - занимают лишь очень малую его часть (у эукариот), а что собой представляет остальной геном - не очень понятно. Предпологается, что помимо многочисленных сигнальных последовательностей в нем есть и просто "мусор", занесенный нам, например, вирусами. На эти участки генома не будет действовать отбор, соответственно мутации не будут отсеиваться.
5)"Для белков есть проверка: сходство структур"
Сходство структур далеко не всегда означает гомологию. Примером могут служить так называемые "белки скользящего зажима" - структуры, которые увеличивают процессивность ДНК-полимераз, обхватывая их и ДНК (и таким образом не давая полимеразе слететь с ДНК). Белки с такой функцией есть и у вирусов, и у бактерий, и у архей, и у эукариот, и все они имеют очень похожую структуру (что объясняется четко заданными необходимыми свойствами, а также крайней важностью белка, которая отсеивает все хоть сколько-нибудь неудачные мутации). Однако далеко не все они являются гомологами:"The T4 bacteriophage also uses a sliding clamp, called gp45 that is a trimer similar in structure to PCNA but lacks sequence homology to either PCNA or the bacterial beta clamp" - [1]. Гомологами также не являются beta clamp бактерий и PCNA:"Even though the E. coli sliding clamp (beta) has only two subunits and shares little sequence homology with the eukaryotic sliding clamp, the overall structures of the beta-clamp and PCNA are quite similar" - [2].
Источники:
НАЗАД ➜ |
| © <Рюмина Екатерина>, 2017 |