Анализ транскриптомов.
1. Подготовка
Были скопированы в рабочую директорию одноконцевые чтения, картирующиеся на участок десятой хромосомы человека (получены путем секвенирования РНК): /P/y14/term3/block4/SNP/rnaseq_reads/chr10.1.fastq и /P/y14/term3/block4/SNP/rnaseq_reads/chr10.2.fastq.
2. Анализ качества и очистка чтений.
С помощью программы FastQC (установлена на kodomo) был проведен анализ качества чтений (результат: для первой реплики и для второй реплике):
| fastqc chr10.1.fastq
| fastqc chr10.2.fastq
Очистка чтений не производилась, поскольку у ридов небольшой разброс длины и приличное качество на всей длине рида.
Ниже приведены некоторые графические данные о этих чтениях: качество нуклеотидов в ридах, качество ячеей иллюмины (приведен потому, что его не было в прошлом практикуме) и те данные, напротив которых стоял восклицательный знак (т. е. данные, показывающие отклонение от нормальных). Для сравнения можно посмотреть "хорошие" данные, приведенные на сайте FastQC.
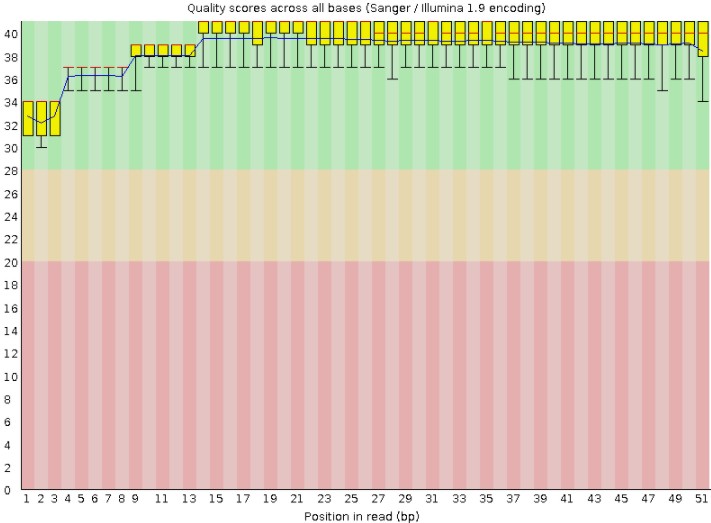 Рисунок 1. Качество чтения нуклеотидов: первая реплика |  Рисунок 2. Качество чтения нуклеотидов: вторая реплика |
По этим данным видно, что все чтения обеих реплик очень приличные: все попдадают в зеленую область (даже вместе с "усами"). Длина ридов небольшая - до 51 нуклеотида (в первой реплике 15462 ридов с длинами 28-51, во второй 11564 ридов с длинами 29-51). Так что очистка ридов в данном случае не нужна. | |
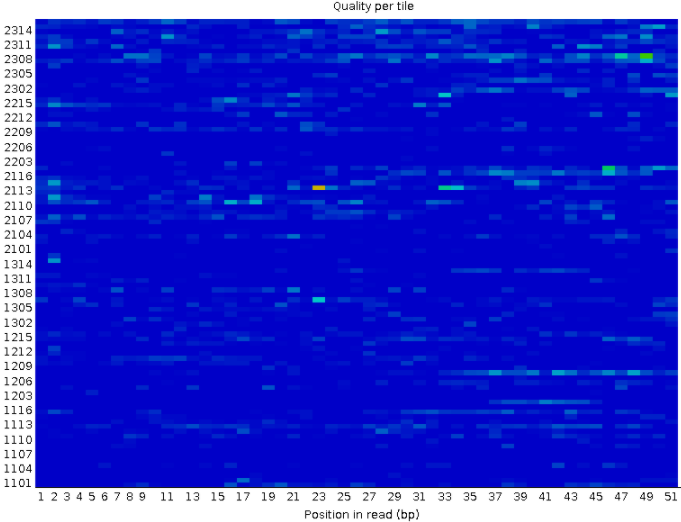 Рисунок 3. Качество ячеек Иллюмины: первая реплика | 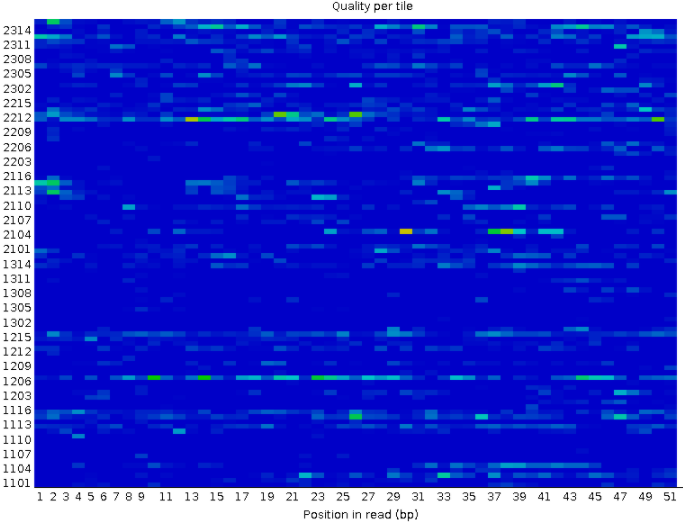 Рисунок 4. Качество ячеек Иллюмины: вторая реплика |
По оси ординат расположены номера ячеек, по оси абсцисс - номер нуклеотида в риде. Качество сигнала в ячейке тем лучше, чем "холоднее" цвет, поэтому идеальной картинкой являлся бы чисто синий фон. Совсем плохо, если есть красные, таких нет. Видно, что часто проблемы если есть, то во многих нуклеотидах одной ячейки, особенно хорошо это заметно во ворой реплике - ячейки 1206 и 2212, в которых очень много плохо прочитанных нуклеотидов. | |
 Рисунок 5. Доля каждого основания на позиции: первая реплика |  Рисунок 6. Доля каждого основания на позиции: вторая реплика |
По распределению доли конкретного нуклеотида на конкретной позиции в риде видно, что помимо странного начала (с пиками цитозина и гуанина и почти отсутсвующих аденина и тимина на первой позиции и дальнейшими странными пиками) в районе 6-9 позиций ридов есть пик тимина, где его процент сильно превосходит процент остальных нуклеотидов (примерно 55% тимина против примерно 20% остальных). Да и на всем остальном протяжении ридов тимина больше, чем любого другого нуклеотида. И если у=цитозина и гуанина еще примерно поровну, то аденина почти везде больше, чем гуанина или цитозина. Понятно, что такая картина воспринимается программой как ненормальная. Причем такой пик наблюдается на чтениях обоих реплик, что указывает либо на систематическую ошибку методики, либо на действительно имеющее место явление. | |
 Рисунок 7. Распределение GC: первая реплика |  Рисунок 8. Распределение GC: вторая реплика |
Реальное распределение GC должен примерно совпадать с теоретическим распределением, однако в данном случае в обеих репликах это не так. Наблюдается, во-первых, сдвиг реального распределения вправо относительно теоретического (увеличение доли GC начинается на более поздних позициях), во-вторых, большой пик в 37-40 позициях. | |
 Рисунок 9. Повторение последовательностей: первая реплика | 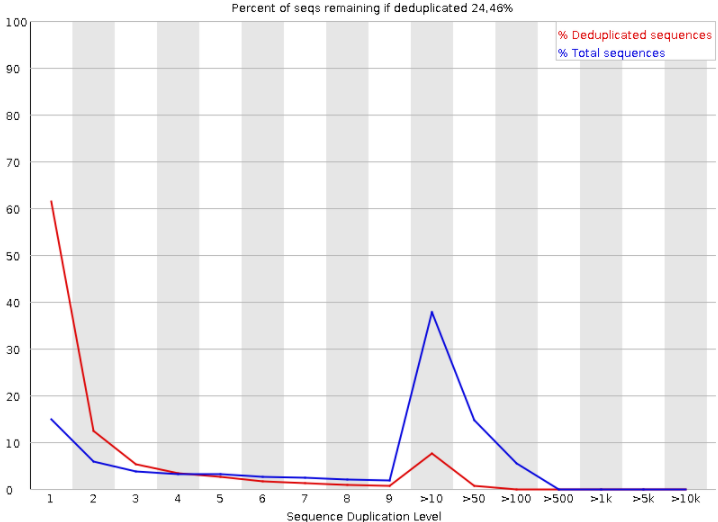 Рисунок 10. Повторение последовательностей: вторая реплика |
Этот график показывает колчество дуплицированных последовательностей, причем синяя линия показывает все количество, а красная - изначальных разных образцов, с которых дуплицировалось все, показанное синей линией. Видно, что в районе 10 наблюдается большой пик не только синий, но и красный, что свидетельствует о большом числе изначальных образцов. Что странно, и даже позволяет предположит контоминацию образца. | |
3. Картирование чтений.
Дальнейшие действия - картирование очищенных чтений по референсной последовательности с помощью программы Hisat2.
Сначала была извлечена необходимая программа:
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
Индексация референсной последовательности балы взята из прошлого практикума (8 файлов - chr10.1.ht2 <...> chr10.8.ht2). Так что следующим этапом сразу было построение выравнивания - такое же, как и в предыдущем практикуме с одним различием: был убран параметр --no-spliced-alignment, запрещавший разрывы в выравнивании (что логично, поскольку транскриптом не обязан быть непрерывным). Также приведены данные, выданные этой программой: число невыровненных ридов, ридов, выровненных один раз и ридов, выровненных больше одного раза.
| hisat2 -x chr10 -U chr10.1.fastq --no-softclip -S chr10.1.sam
15462 reads; of these:
15462 (100.00%) were unpaired; of these:
206 (1.33%) aligned 0 times
15194 (98.27%) aligned exactly 1 time
62 (0.40%) aligned >1 times
98.67% overall alignment rate
| hisat2 -x chr10 -U chr10.2.fastq --no-softclip -S chr10.2.sam
11564 reads; of these:
11564 (100.00%) were unpaired; of these:
160 (1.38%) aligned 0 times
11367 (98.30%) aligned exactly 1 time
37 (0.32%) aligned >1 times
98.62% overall alignment rate
4. Анализ выравнивания
С помощью пакета samtools был произведен анализ выравнивания.
Сначала файлы с выравниванием ридов по референсной последовательности (chr10.*.sam) были переведены в бинарный формат (chr10.*.bam):
| samtools view chr10.1.sam -o chr10.1.bam -b
| samtools view chr10.2.sam -o chr10.2.bam -b
Затем риды в этом файле были отсортированы по своей левой (начальной) координате в референсе (файл *_temp.txt - временный файл, необходимый для работы программы, файл chr10.*_sort.bam - вывод программы):
| samtools sort chr10.1.bam -T chr10.1_temp.txt -o chr10.1_sort.bam
| samtools sort chr10.2.bam -T chr10.2_temp.txt -o chr10.2_sort.bam
получившийся файл был проиндексирован:
| samtools index chr10.1_sort.bam
| samtools index chr10.2_sort.bam
5. IGV
Как ив предыдущем практикуме, в программу IGV были загружены файлы .bam. На рисунках 11 приведено чуть ли не единственное место, где были риды (верхний трек соответствует транскриптому, нижний - секвенированию ДНК из предыдущего практикума). Интересно, что в этом месте отсутствуют риды предыдущего практикума. Для сравнения на рисунке 12 приведено место с наибольшим покрытием хромосомы секвенированием ДНК из прошлого практикума.
Помимо разности в покрытии хромосомы (у трансрикптома оно почти никакое), видна и разница в выравнивании ридов, а именно в наличии "разрывов" в ридах транскриптома (на рисунке 11 они показаны горизонтальными голубоватыми линиями, соединяющими куски ридов). Понятно, что такие разрывы получаются из-за сплайсинга.
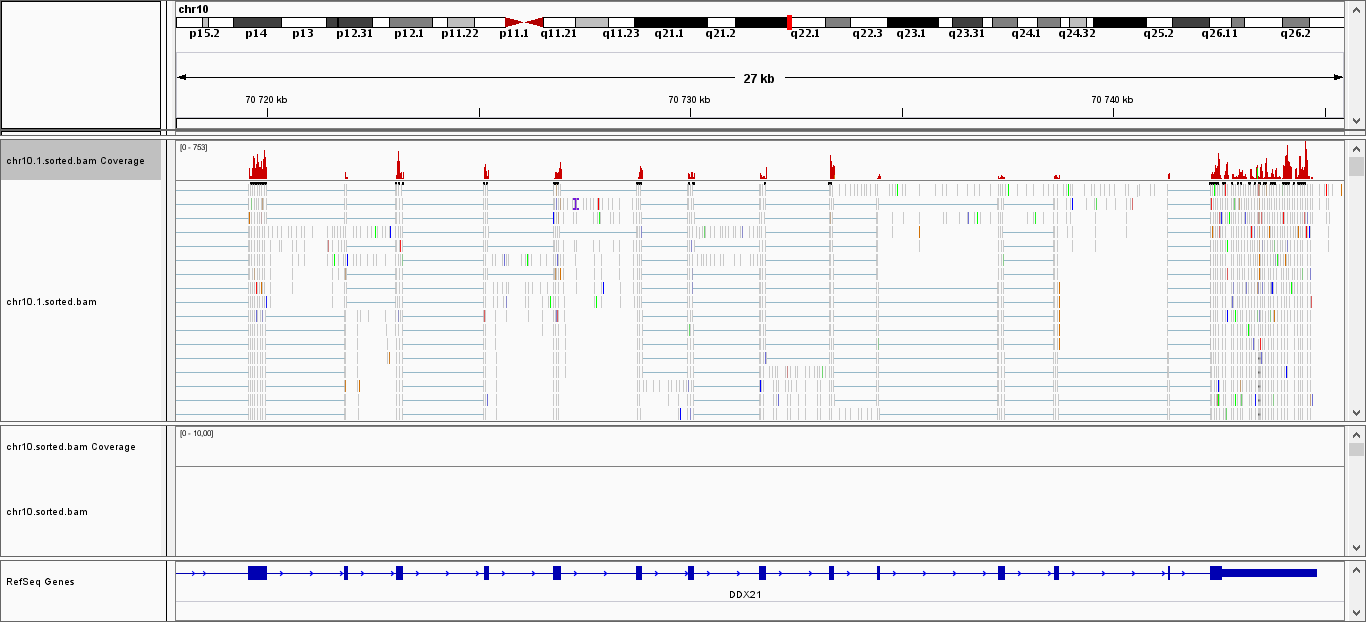 Рисунок 11. |
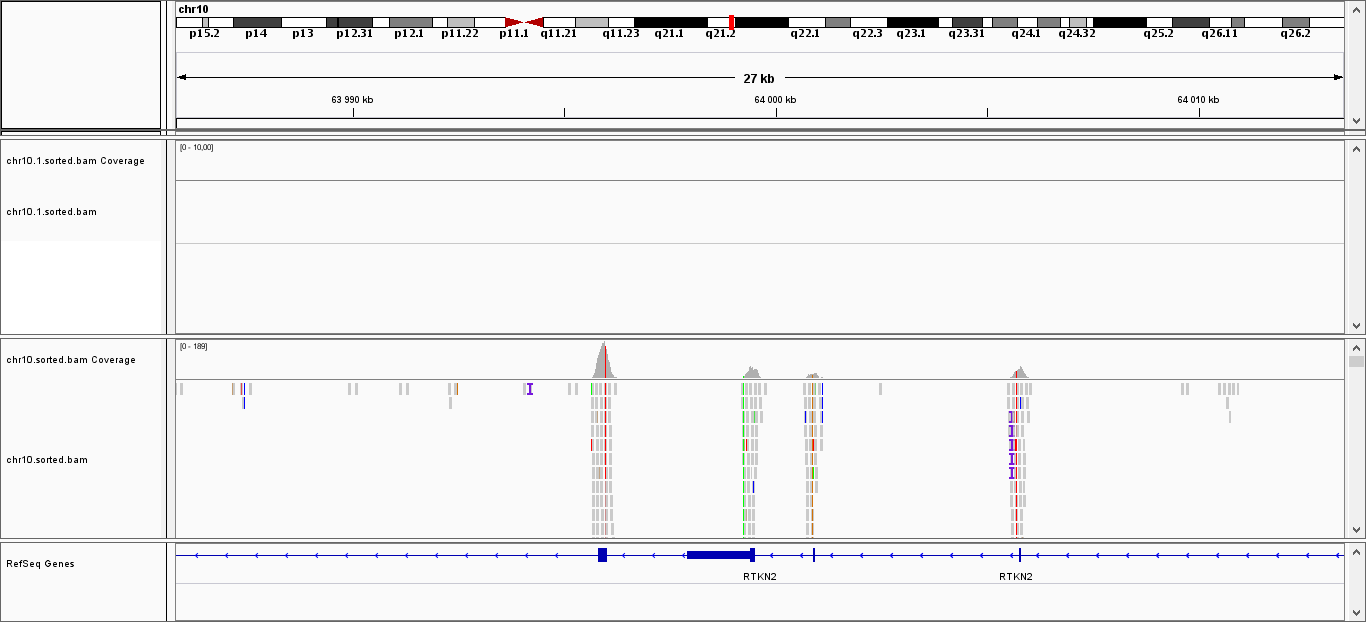 Рисунок 12. |
6. Bedtools
Чтобы узнать, какие гены окаались покрыты ридами и сколькими ридами, был использован пакет bedtools. Сначала файлы .bam быди переведены в соответствующий формат:
| bedtools bamtobed -i chr10.1_sort.bam > chr10.1.bed | bedtools bamtobed -i chr10.2_sort.bam > chr10.2.bedЗатем был экспортирован сам покет программ:
| export PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin
После чего с помощью команды intersect производилось сравнение между файлом с разметкой генов по версии Gencode для генома человека сборки hg19 (gencode.genes.bed) и файлами с ридами. Параметры -a задают файл, записи из которого сравниваются с другим файлом (-b). В данной ситуации файлом -a была разметка, т. к. именно в ней приведены названия генов. Параметр -c выводит
Но в этом файле содержатся гены всех хромосом, поэтому с помощь grep были извлечены строки, содержащие 'chr10'. В получившимся файле строки были отсортированы, после чего нулевые строки были удалены. Результат можно видеть в таблице 1.
| bedtools intersect -a gencode.genes.bed -b chr10.2.bed -c | grep 'chr10' > chr10.2_inter.bed | bedtools intersect -a gencode.genes.bed -b chr10.1.bed -c | grep 'chr10' > chr10.1_inter.bed
В таблице 1 я удалила полностью совпадающие строки и проставила номера экзонов там, где они согласуются с таковыми в IGV (см. рис. 13). Смысл остальных участков мне не совсем ясен, т. к. они то пересекаются друг с дргом, то состоят всего из двух нуклеотидов. Поэтому я дальше буду опираться на совпадающие с IGV куски.
| Реплика 1 | Реплика 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
Таблица 1. Покрытие генов.
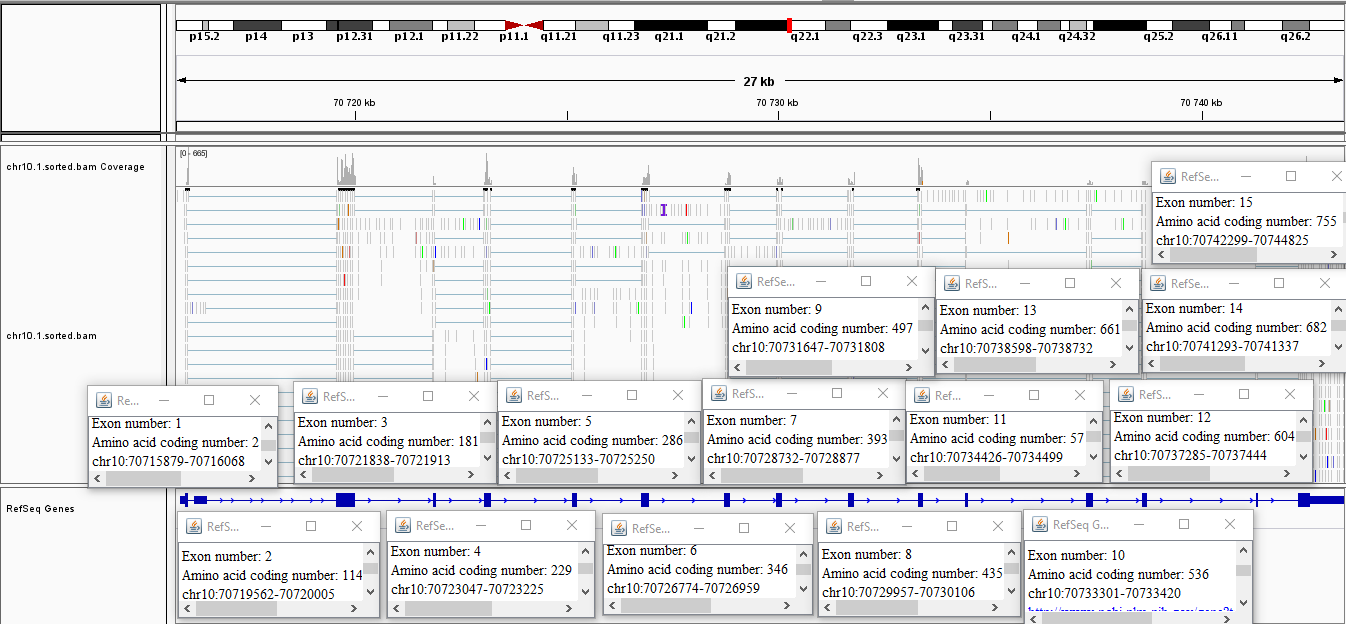
Рисунок 13. Разметка экзонов гена DDX21 согласно IGV.
Как бы то ни было, таблица 1 позволяет понять, что найден транскрипт только одного известного гена (DDX21), а также "miscellaneous RNA" - то есть непонятно какая РНК. Найдя этот ген с помощью IGV, можно узнать число экзонов. IGV также приводит ссылку на описание этого гена на NCBI.
- Описание гена DDX21 (DExD-box helicase 21):
- Координаты: 70715884 - 70744829 (по intersect); 70715879 - 70744825 по IGV.
- Число экзонов: IGV показывает 15, но на NCBI написано 16.
- Функция: предположительно, РНК-хеликазы. Участвуют в процесах, включающих изменение вторичной структуры РНК, таких как инициация трансляции, сплайсинг, сборка рибосом и сплайсосом.
7. Работа с bedtools
- Получение из файла с выравниванием файла с чтениями в формате fastq. chr10.1.fq
| bedtools bamtofastq -i chr10.1_sort.bam -fq chr10.1.fq
- Получение файла с нуклеотидной последовательностью (.fasta) для гена DDX21 (экзон 2) - последовательность взята из fasta файла с последовательностью 10-ой хромосомы..
| grep '70741293' chr10.1_inter.bed | head -1 > test.bed | bedtools getfasta -fi chr10.fasta -bed test.bed >chr10:70741293-70741337 TGCTTTGATTTCTAGGGTGTTTGCTTTGATGTACCTACCGCATC
- Объединение чтений в кластеры (параметр -d меняет максимальное расстояние, пр котором риды еще считаются одним кластером. Т. к. у меня получилось 23 кластера при нулевом расстоянии, я не меняла этот параметр. Более того, с помощью merge я узнала, что уже при расстоянии в 230 п. н. все влипается в 1 кластер (для этого проще использовать merge, потому что все объединяет в одну запись и это смотреть быстрее). clust.bed
| bedtools cluster -i chr10.1.bed -d 0 > clust.bed | bedtools merge -i chr10.1.bed -d 230 chr10 70714614 70746458 | bedtools merge -i chr10.1.bed -d 220 chr10 70714614 70746178 chr10 70746407 70746458
- Набор 1000 случайных фрагментов по 200 нуклеотидов из хромосомы. Эта программа требует файл genome. Необходимая информация о моей 10 хромосоме была мною получена отсюда (из скачанного файла, ссылка на которой на этой странице). Потом она была записана в файл chr10.genome. Сначала был получен файл с координатами данных фрагментов - random.bed, затем эти фрагменты были вырезаны их файла с последовательностью хромосомы (chr10.fasta) - pieces.bed.
| bedtools random -n 1000 -l 200 -g chr10.genome > random.bed | bedtools getfasta -fi chr10.fasta -bed random.bed > pieces.bed
- Получение координат экзона 2 гена DDZX2, расширенных на 1000 нуклеотидов в обе стороны.
| bedtools slop -i test.bed -g chr10.genome -b 1000 chr10 70740293 70742337 protein_coding DDX21 130
- Получение координат экзона 2 гена DDZX2, сдвинутых на 500 нуклеотидов ближе к началу хромосомы.
| bedtools slop -i test.bed -g chr10.genome -l 500 -r -500 chr10 70740793 70740837 protein_coding DDX21 130
- Получение файла с координатами интервалов, покрытых чтениями, с информацией о покрытии (двух последних столбца файла): число записей в gencode.genes.bed, пересекающихся с данным ридом, и покрытие (доля). an.bed
| bedtools annotate -i chr10.1.bed -files gencode.genes.bed -both > an.bed
