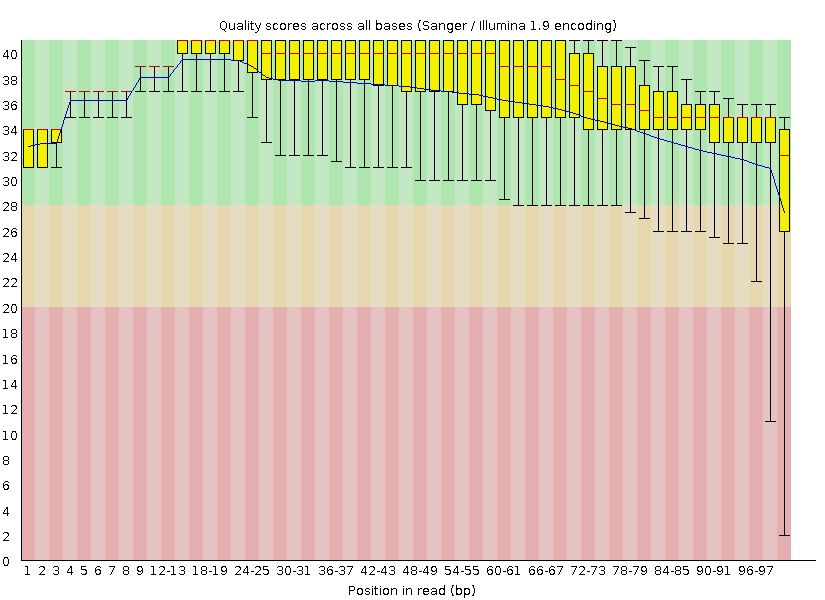
Рис1. До чистки Trimmomatic (8367 ридов).
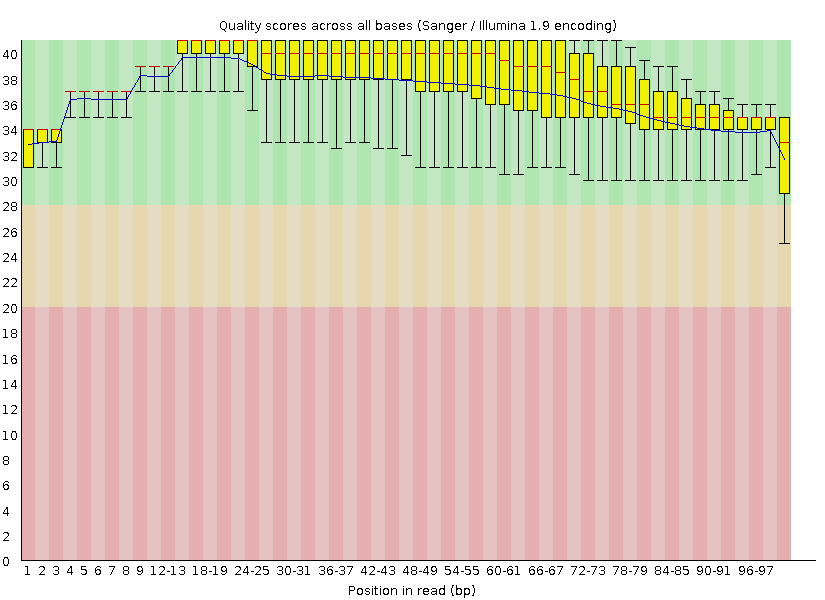
Рис2. После чистки Trimmomatic (8227 ридов).
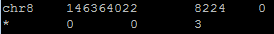
Рис3. Количество картированных (8224) и некартированных (0) на хромосому чтений.
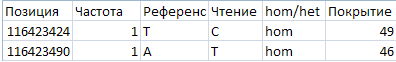
Рис4. Два полиморфизма и индель.
| Команда |
Функция |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp2.vcf > conv_snp.avinput |
конвертирует .vcf файл в формат, удобоваримый для annovar |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 conv_snp.avinput -outfile refgeneann /nfs/srv/databases/annovar/humandb/ |
аннотация файла со списком замен по базе RefGene |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 conv_snp.avinput -outfile snp138ann /nfs/srv/databases/annovar/humandb/ |
аннотация файла со списком замен по базе SNP138 |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 conv_snp.avinput -outfile 1000genomesann /nfs/srv/databases/annovar/humandb/ |
аннотация файла со списком замен по базе 1000 genomes |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 conv_snp.avinput -outfile gwasann /nfs/srv/databases/annovar/humandb/ |
аннотация файла со списком замен по базе GWAS |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 conv_snp.avinput -outfile clinvarann /nfs/srv/databases/annovar/humandb/ |
аннотация файла со списком замен по базе Clinvar |
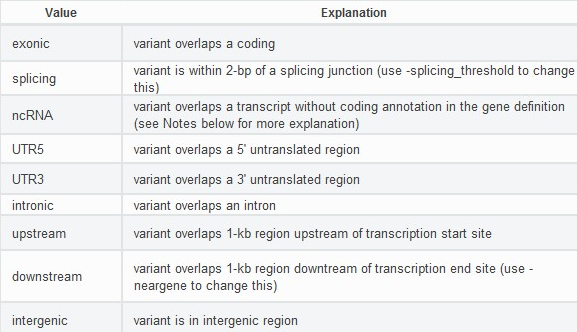
Рис5. Категории полиморфизмов в аннотации RefGene.
Рис6. Две самые частые замены.