Также требовалось проделать аналогичную работу с набором контигов той же бактерии из метагенома кишечника человека. Результат доступен по ссылке. в этот раз избегаемых сайтов со значением контраста было уже 4:
Можно заметить, что у бактерии из метагенома кишечника человека присутствуют оба сайта, найденные в геноме бактерии из генбанка. Это может означать, что бактерия, геном которой использовался в первом случае, недавно потеряла соответствующие рестриктазы.
Последовательность Шайна-Дальгарно - сайт связывания рибосом на молекуле мРНК прокариот, обычно на расстоянии около 10 нуклеотидов до стартового кодона AUG. Консенсусом является последовательность из шести нуклеотидов AGGAGG. Комплементарное взаимодействие между последовательностями Шайна-Дальгарно и анти-Шайна-Дальгарно (консенсус CCUCCU, расположена на 3'-конце молекулы 16S рибосомной РНК) служит для помещения старт-кодона мРНК в P-сайт рибосомы для начала биосинтеза белка.
При выполнении задания я искал такие последовательности в геноме археи Methanosaeta concilii GP6, выданной мне в первом семестре. Для этого я проделал следующие шаги:
К сожалению, поиск не принёс результата: МЕМЕ не нашел ни одного мотива. Поэтому для дальнейшего выполнения задания была выбрана другая бактерия - Aquifex aeolicus, последовательность хромосомы и таблица особенностей также были взяты со страницы бактерии в БД NCBI Assembly. C полученными данными была проделана та же самая работа, что и раньше. Для первичного анализа и поиска мотива были выбраны 400 самых длинных кодирующих последовательностей, поиск мотива велся на расстоянии -16 до старт кодона. Получив файл, содержащий последовательности с указанными координатами, я использовал его для поиска с помощью МЕМЕ со следующими параметрами: длина мотива от 4 до 10 нуклеотидов, встречается 0 или 1 мотив, поиск только на данной цепи.
На этот раз МЕМЕ нашел искомый мотив и составил нужную нам позиционно-весовую матрицу (PWM), которая дальше была использована для поиска этого мотива в upstream-области всех генов. Этот поиск был проведен с помощью сервиса FIMO. В этот раз область поиска была расширена до -25 нуклеотидов от старт-кодона, порог e-value задан 0.01. Последовательность Шайна-Дальгарно была найдена примерно в 80% всех генов, что является очень даже неплохим результатом. Итог работы сервиса - таблицу в формате Excel можно скачать здесь. Лого мотива приведено на рисунке 2:

Для работы получил файл chipseq_chunk14.fastq, содержащий риды Illumina одного из участков хромосомы человека. Сначала был проведен контроль качества ридов с
помощью команды fastqc chipseq_chunk14.fastq. Отчёт о проделанной командой работе содержится в файле fastqc_report.html. Всего ридов 6546. Kачество чтения очень высокое на всём
протяжении последовательности, поэтому программа Trimmomatic для очистки чтений от участков плохого качества не применялась.
Прочтения были картированы на проиндексированный геном человека:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk14.fastq > chipseq_chunk14.sam
samtools view -b chipseq_chunk14.sam -o aln.bam- перевод в бинарный формат;
samtools sort aln.bam out.predix.bam- сортировка по координате в референсе начала чтения;
samtools index out.predix.bam- индексация полученного файла;
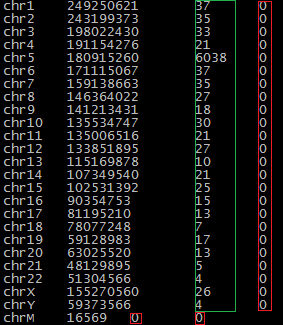
samtools idxstats out.predix.bam- узнаем, сколько ридов было откартировано, см рис 3.1.
macs2 callpeak -n chunk14 -t chipseq_chunk14.sorted.bam --nomodel
track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 14"
browser position chr5:77779600-78440400
Файл был подан на вход форме custom tracks в UCSC Genome Browser. Скриншот - на рис.3.2:
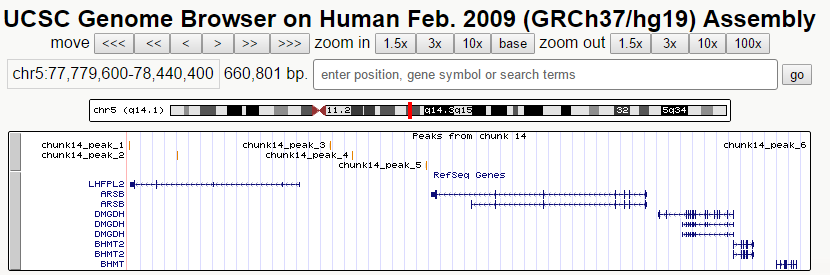
Рассмотрим подробнее пики 1 и 2.
| Пик | 1 | 2 |
| Длина пика | 284 | 283 |
| -LOG10(pvalue) | 13.17144 | 21.30753 |
| Вершина пика относительно начала | 92 от начала пика | 144 от начала пика |
| Положение в геноме | Оба пика пересекают экзоны гена LHFPL2, кодирующего трансмембранный белок lipoma HMGIC fusion partner-like 2 protein, мутации в котором приводят к глухоте. | |
Чем больше величина -LOG10(pvalue), тем меньше числовое значение pvalue и, следовательно, тем достовернее пик. Таким образом, второй пик является самым достоверным из всех пяти.
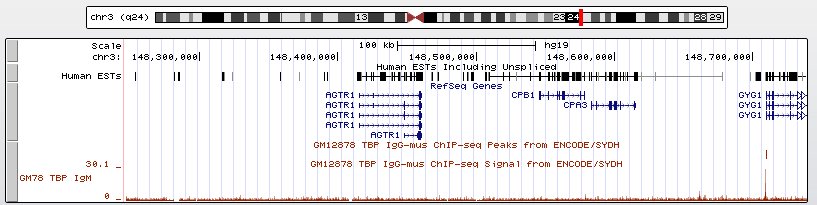
TATA-бокс связывающий фактор TBP - архейный и эукариотический белок, узнающий восьминуклеотидный сигнал в ДНК с консенсусом TATAWAAR. Он является одним из ключевых ДНК-узнающих белков при образовании на промоторе генов комплекса TFIID инициации транскрипции с помощью Pol II. Тем не менее, лишь часть промоторов имеет сигнал TATA-box, связываемый TBP. Цель этого задания - найти в геноме человека три гена, имеющих такой сигнал, и один ген, не имеющий сигнала. Поиск велся с помощью сервиса UCSC GenomeBrowser. Для белка TBP (TATA binding protein) был выбран ChiP-Seq-эксперимент, проведенный на клеточной линии GM12878. В эксперименте использовались антитела к TBP ab62126.
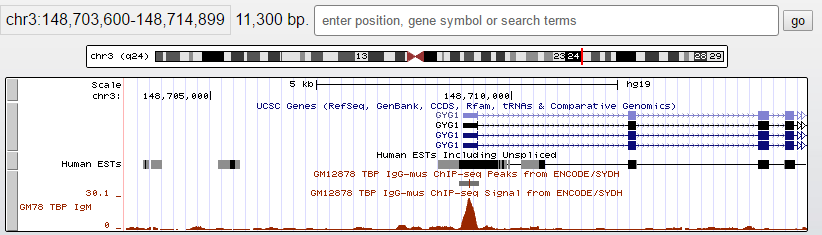
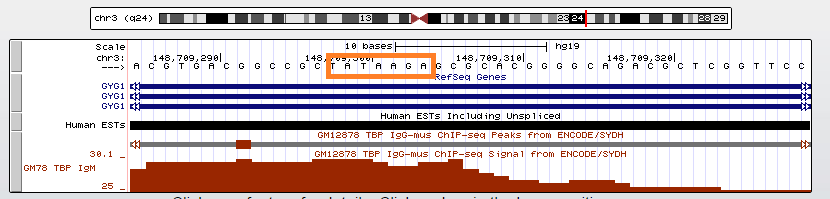
Первый ген, имеющий пик - ген GYG1, кодирующий белок гликогенин-1 - гликозилтрансферазу, катализирующую формирование небольших глюкозных полимеров. Размер кодирующей последовательности составляет 35351 нуклеотидов. Позиция в геноме - 148709428-148744778, на 3 хромосоме (+ - цепь). Ген имеет 6 экзонов.
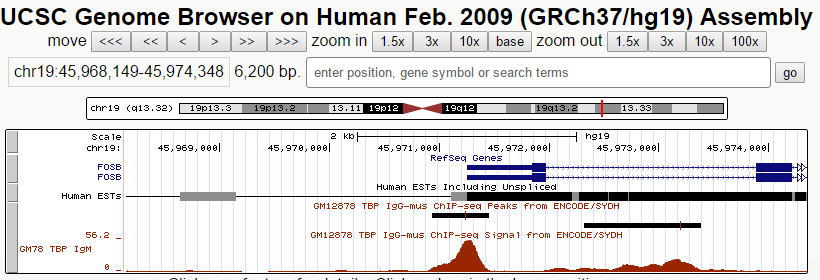
Второй ген, у которого нашелся пик - ген FOSB. Семейство генов Fos включает в себя 4 гена: FOS, FOSB, FOSL1 и FOSL2. Они кодируют белки - лейциновые молнии, которые учавствуют в димеризации белков семейства JUN, формируя комплекс - транскрипционный фактор AP-1. Размер кодирующей последовательности гена - 7185 нуклеотидов. В геноме расположен на "+"-цепи 19 хромосомы на позиции 45971253-45978437. Имеет 3 экзона.
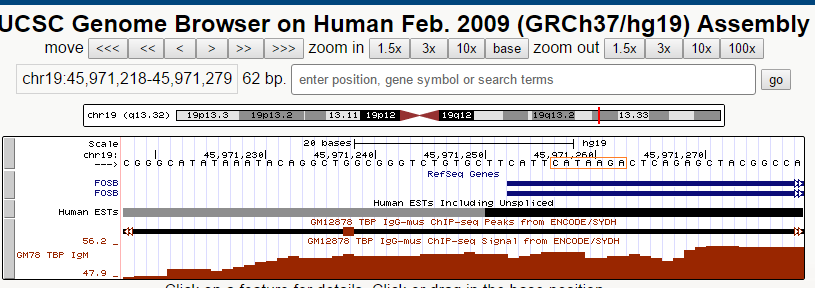
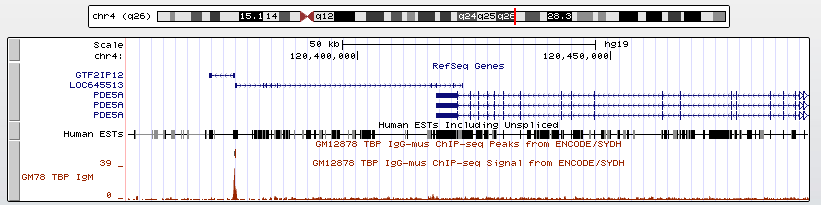
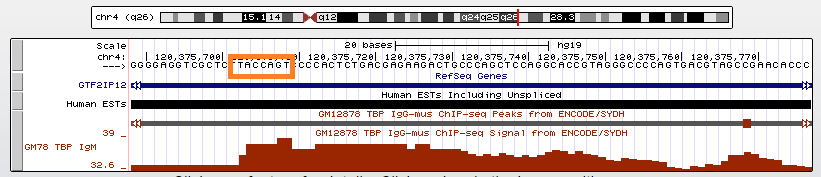
Третий ген с выраженным пиком - PDE5A, кодирующий ГМФ-связывающую фосфодиэстеразу. Положение в геноме - 120415550-120549981 на "+" - цепи 4 хромосомы. Длина гена - 7005 нуклеотидов.
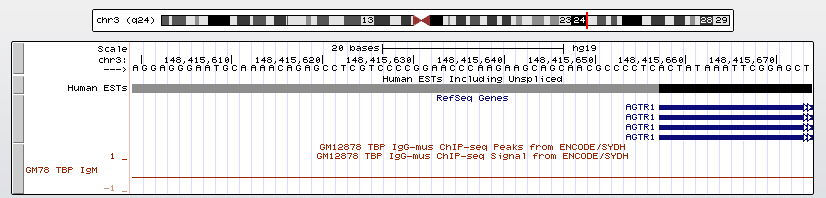
В качестве примера гена без ТВР-связывающего мотива был выбран ген AGTR1. Его продукт - рецептор ангиотензина-2 первого типа. Находится на "+" - цепи третьей хромосомы. Положение на хромосоме - 148415658-148460790. Длина гена - 2272 нуклеотида.