Практикум 11
Ссылка на поиск в SwissProt
Ссылка на выравнивание двух последовательностей
Задание 1
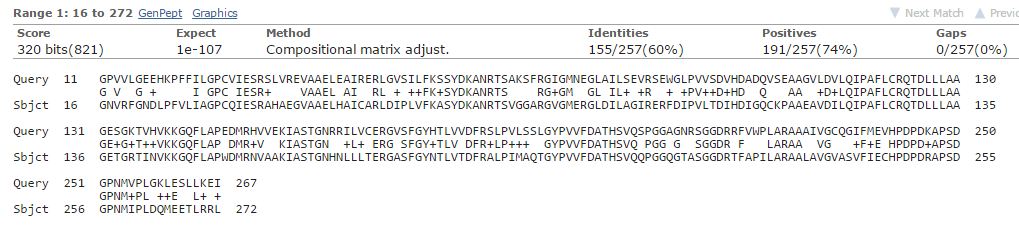
В этом задании был запущен BLAST с идентификатором WP_014961570.1 в Swissprot. Было найдено 248 последовательностей.| Лучшая находка | Худшая находка | Находка из центра | |
| Идентификатор находки | Q0BTX5.1 | P40332.2 | Q83FU2.3 |
| Score | 320 | 32 | 225 |
| Идентичные остатки,% | 60 | 25 | 43 |
| Сходные остатки,% | 74 | 45 | 62 |
| E-value | 1e-107 | 4.7 | 3e-70 |
Выравнивания:
Лучшее выравнивание:
Худшее выравнивание:
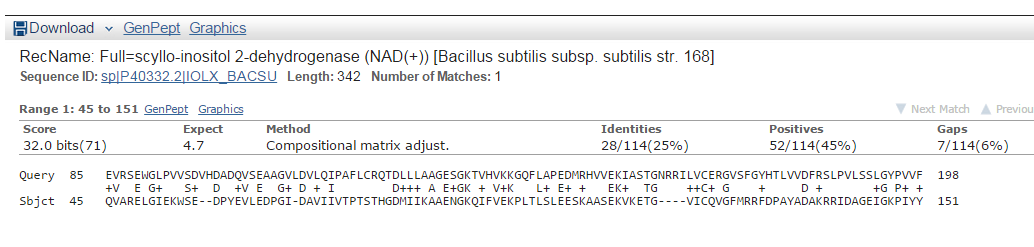
Выравнивание из центра:
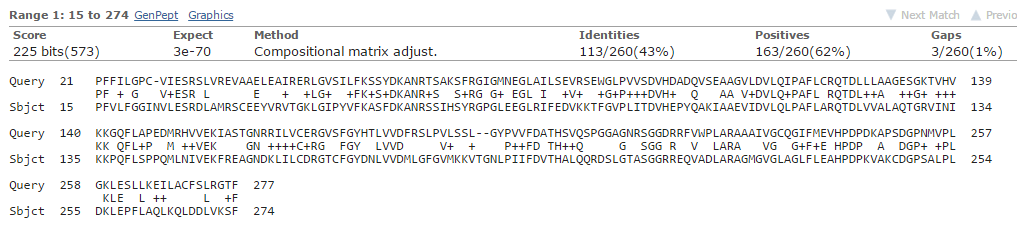
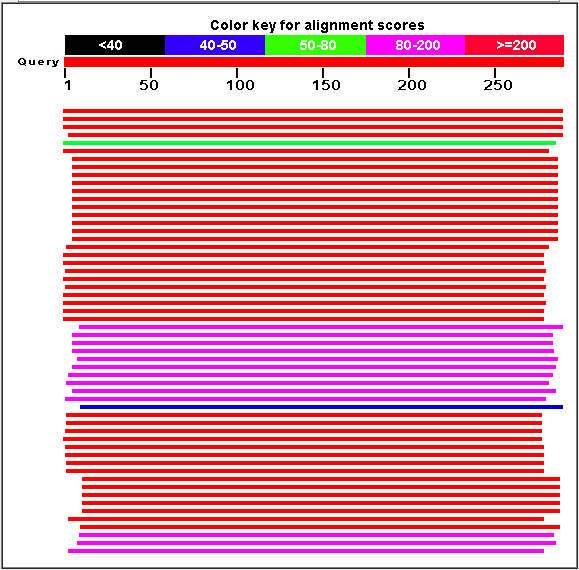
Гомологами исходной последовательности (согласно приведённым в задании данным: E-value <= 0.001, Query cover >= 70%) можно считать 237 последовательностей.
Графическое представление результатов:
Задание 2
Все без исключения находки принадлежат таксономической группе eubacteria. Поэтому при поиске только по группе eubacteria не изменилось ни число находок(в т.ч. и достоверных), ни Query cover. Для примера рассимотрим находку с идентификатором P39912.1 (худшая находка):Рисунок 1. Поиск по всем таксонам
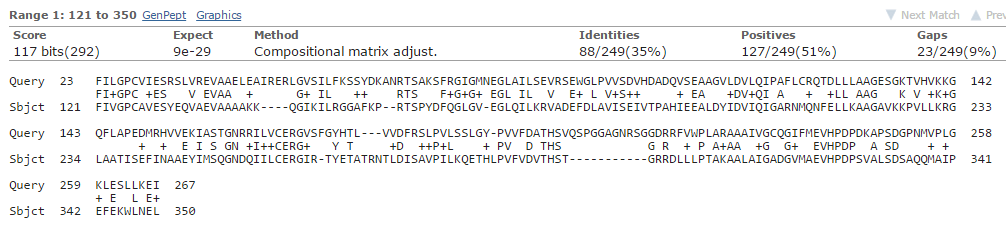
Рисунок 2. Поиск по eubacteria
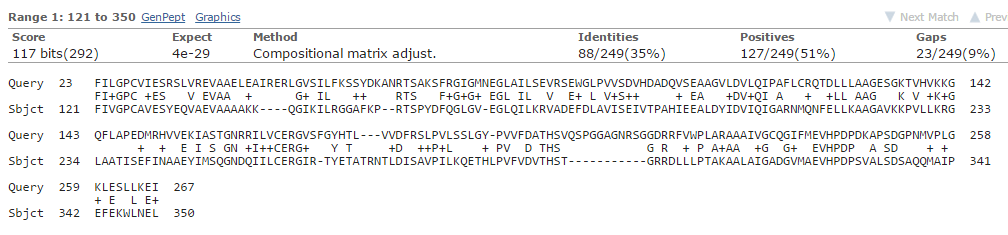
Выравнивания получились одинаковые. Score не изменился, т.к. матрица и последовательности остались прежними. E-value уменьшился, т.к. уменьшился размер банка (чем меньше банк, тем меньше E-value).
Задание 3
Было произведено выравнивание двух последовательностей: исходной (WP_014961570.1) и худшей (WP_014961570.1). Ниже представлено исходное выравнивание: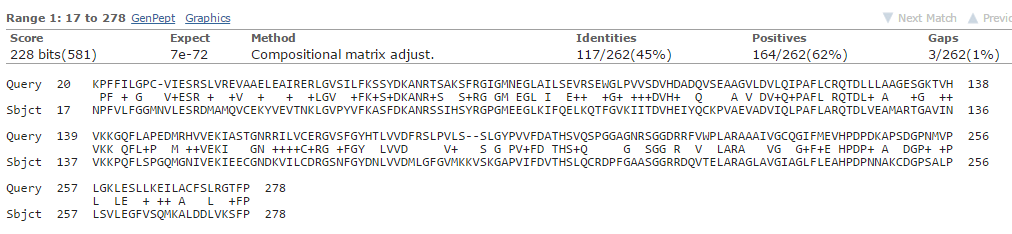
И выравнивание двух последовательностей:
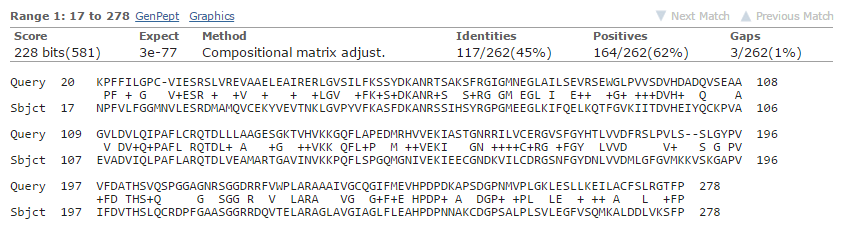
Карта локального сходства:
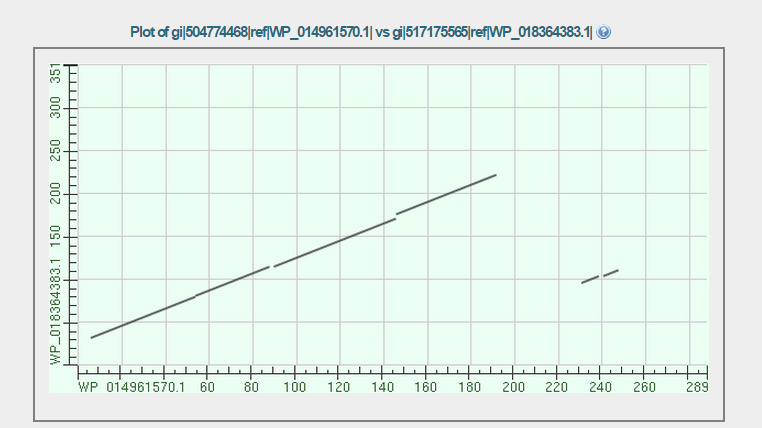
Сплошные участки на графике соответствуют участкам выравнивания без гэпов. Например, участки с 20 по 85 и с 90 по 145 в query. Учитывая тот факт, что была взята "худшая" из гомологичных находок, можно предположить, что данные участки играют некоторую функциональную роль.
Задание 4
В качестве базы данных был использован файл align_03.fasta из 8 практикума.Лучшая находка:
Score = 18.1 bits (35), Expect = 0.60, Method: Compositional matrix adjust.
Identities = 7/19 (37%), Positives = 9/19 (47%), Gaps = 0/19 (0%)

О гомологии последовательностей говорить не приходится. Во-первых, мало positives и identities по сравнению с заведомо гомологичными выравниваниями; во-вторых, score довольно мал (притом всё вышесказанное справедливо не для всей последовательности, а лишь для лучшего её участка). E-value велик, несмотря на малый размер БД.