Изучение работы методов контроля температуры в молекулярной динамике
import numpy as np
import matplotlib.pyplot as plt
import time
%matplotlib inline
Получим файл с координатами молекулы этана:
! wget http://kodomo.fbb.msu.ru/FBB/year_08/term6/etane.gro
--2019-04-21 12:49:38-- http://kodomo.fbb.msu.ru/FBB/year_08/term6/etane.gro Resolving kodomo.fbb.msu.ru... 192.168.180.1 Connecting to kodomo.fbb.msu.ru|192.168.180.1|:80... connected. HTTP request sent, awaiting response... 200 OK Length: 399 Saving to: `etane.gro.1' 100%[======================================>] 399 --.-K/s in 0s 2019-04-21 12:49:38 (31.1 MB/s) - `etane.gro.1' saved [399/399]
Файл топологии для этана:
! cat et.top
#include "/usr/share/gromacs/top/oplsaa.ff/forcefield.itp"
[ moleculetype ]
; Name nrexcl
et 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass
1 opls_139 1 ETH C1 1 -0.189 12.01
2 opls_139 1 ETH C2 2 -0.155 12.01
3 opls_140 1 ETH H1 3 0.0059 1.008
4 opls_140 1 ETH H2 4 0.0059 1.008
5 opls_140 1 ETH H3 5 0.0059 1.008
6 opls_140 1 ETH H4 6 0.0056 1.008
7 opls_140 1 ETH H5 7 0.0056 1.008
8 opls_140 1 ETH H6 8 0.0056 1.008
[ bonds ]
; ai aj funct b0 kb
1 2 1
1 3 1
1 4 1
1 5 1
2 6 1
2 7 1
2 8 1
[ angles ]
; ai aj ak funct phi0 kphi
;around c1
3 1 4 1
4 1 5 1
3 1 5 1
2 1 3 1
2 1 4 1
2 1 5 1
;around c2
6 2 7 1
7 2 8 1
6 2 8 1
1 2 6 1
1 2 7 1
1 2 8 1
[ dihedrals ]
; ai aj ak al funct
3 1 2 6 3
3 1 2 7 3
3 1 2 8 3
4 1 2 6 3
4 1 2 7 3
4 1 2 8 3
5 1 2 6 3
5 1 2 7 3
5 1 2 8 3
[ pairs ]
; список атомов 1-4
; ai aj funct
3 6
3 7
3 8
4 6
4 7
4 8
5 6
5 7
5 8
[ System ]
; any text here
first one
[ molecules ]
;Name count
et 1
Дано 5 конфигурационных файлов с разными алгоритмами контроля температуры: * be.mdp - метод Берендсена для контроля температуры. * vr.mdp - метод "Velocity rescale" для контроля температуры. * nh.mdp - метод Нуза-Хувера для контроля температуры. * an.mdp - метод Андерсена для контроля температуры. * sd.mdp - метод стохастической молекулярной динамики.
methods = ['be', 'vr', 'nh', 'an', 'sd']
for i in methods:
# скачаем конфигурационные файлы
!wget http://kodomo.fbb.msu.ru/FBB/year_08/term6/{i}.mdp
# построим входные файлы для молекулярно-динамического движка mdrun
!grompp -f {i}.mdp -c etane.gro -p et.top -o et_{i}.tpr
#запустим mdrun
!mdrun -deffnm et_{i} -v -nt 1
#конвертируем в pdb
!trjconv -f et_{i}.trr -s et_%s.tpr -o et_%s.pdb
--2019-04-21 12:49:46-- http://kodomo.fbb.msu.ru/FBB/year_08/term6/be.mdp
Resolving kodomo.fbb.msu.ru... 192.168.180.1
Connecting to kodomo.fbb.msu.ru|192.168.180.1|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 1356 (1.3K)
Saving to: `be.mdp.1'
100%[======================================>] 1,356 --.-K/s in 0s
2019-04-21 12:49:46 (175 MB/s) - `be.mdp.1' saved [1356/1356]
:-) G R O M A C S (-:
Glycine aRginine prOline Methionine Alanine Cystine Serine
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) grompp (-:
Option Filename Type Description
------------------------------------------------------------
-f be.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c etane.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p et.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o et_be.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]v bool no Be loud and noisy
-time real -1 Take frame at or first after this time.
-[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
-maxwarn int 0 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
-[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
-[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Ignoring obsolete mdp entry 'title'
Ignoring obsolete mdp entry 'cpp'
Replacing old mdp entry 'unconstrained-start' by 'continuation'
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.5#
NOTE 1 [file be.mdp]:
nstcomm < nstcalcenergy defeats the purpose of nstcalcenergy, setting
nstcomm to nstcalcenergy
NOTE 2 [file be.mdp]:
The Berendsen thermostat does not generate the correct kinetic energy
distribution. You might want to consider using the V-rescale thermostat.
Generated 332520 of the 332520 non-bonded parameter combinations
Generating 1-4 interactions: fudge = 0.5
Generated 332520 of the 332520 1-4 parameter combinations
Excluding 3 bonded neighbours molecule type 'et'
NOTE 3 [file et.top, line 76]:
System has non-zero total charge: -0.309500
Total charge should normally be an integer. See
http://www.gromacs.org/Documentation/Floating_Point_Arithmetic
for discussion on how close it should be to an integer.
Analysing residue names:
There are: 1 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Number of degrees of freedom in T-Coupling group System is 21.00
NOTE 4 [file be.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
This run will generate roughly 8 Mb of data
There were 4 notes
Back Off! I just backed up et_be.tpr to ./#et_be.tpr.1#
gcq#70: "What's Your Definition Of Dirty ?" (G. Michael)
:-) G R O M A C S (-:
GROningen MAchine for Chemical Simulation
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s et_be.tpr Input Run input file: tpr tpb tpa
-o et_be.trr Output Full precision trajectory: trr trj cpt
-x et_be.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi et_be.cpt Input, Opt. Checkpoint file
-cpo et_be.cpt Output, Opt. Checkpoint file
-c et_be.gro Output Structure file: gro g96 pdb etc.
-e et_be.edr Output Energy file
-g et_be.log Output Log file
-dhdl et_be.xvg Output, Opt. xvgr/xmgr file
-field et_be.xvg Output, Opt. xvgr/xmgr file
-table et_be.xvg Input, Opt. xvgr/xmgr file
-tablep et_be.xvg Input, Opt. xvgr/xmgr file
-tableb et_be.xvg Input, Opt. xvgr/xmgr file
-rerun et_be.trr Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi et_be.xvg Output, Opt. xvgr/xmgr file
-tpid et_be.xvg Output, Opt. xvgr/xmgr file
-ei et_be.edi Input, Opt. ED sampling input
-eo et_be.edo Output, Opt. ED sampling output
-j et_be.gct Input, Opt. General coupling stuff
-jo et_be.gct Output, Opt. General coupling stuff
-ffout et_be.xvg Output, Opt. xvgr/xmgr file
-devout et_be.xvg Output, Opt. xvgr/xmgr file
-runav et_be.xvg Output, Opt. xvgr/xmgr file
-px et_be.xvg Output, Opt. xvgr/xmgr file
-pf et_be.xvg Output, Opt. xvgr/xmgr file
-mtx et_be.mtx Output, Opt. Hessian matrix
-dn et_be.ndx Output, Opt. Index file
-multidir et_be Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string et_be Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 1 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Back Off! I just backed up et_be.log to ./#et_be.log.1#
Getting Loaded...
Reading file et_be.tpr, VERSION 4.5.5 (single precision)
Loaded with Money
Back Off! I just backed up et_be.trr to ./#et_be.trr.1#
Back Off! I just backed up et_be.edr to ./#et_be.edr.1#
starting mdrun 'first one'
250000 steps, 250.0 ps.
step 249900, remaining runtime: 0 s
Writing final coordinates.
Back Off! I just backed up et_be.gro to ./#et_be.gro.1#
step 250000, remaining runtime: 0 s
NODE (s) Real (s) (%)
Time: 3.820 4.082 93.6
(Mnbf/s) (MFlops) (ns/day) (hour/ns)
Performance: 0.000 390.670 5654.473 0.004
gcq#164: "When It Starts to Start It'll Never Stop" (Magnapop)
:-) G R O M A C S (-:
Gnomes, ROck Monsters And Chili Sauce
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) trjconv (-:
Option Filename Type Description
------------------------------------------------------------
-f et_be.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-o et_%s.pdb Output Trajectory: xtc trr trj gro g96 pdb
-s et_%s.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-n index.ndx Input, Opt. Index file
-fr frames.ndx Input, Opt. Index file
-sub cluster.ndx Input, Opt. Index file
-drop drop.xvg Input, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-tu enum ps Time unit: fs, ps, ns, us, ms or s
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-skip int 1 Only write every nr-th frame
-dt time 0 Only write frame when t MOD dt = first time (ps)
-[no]round bool no Round measurements to nearest picosecond
-dump time -1 Dump frame nearest specified time (ps)
-t0 time 0 Starting time (ps) (default: don't change)
-timestep time 0 Change time step between input frames (ps)
-pbc enum none PBC treatment (see help text for full
description): none, mol, res, atom, nojump,
cluster or whole
-ur enum rect Unit-cell representation: rect, tric or compact
-[no]center bool no Center atoms in box
-boxcenter enum tric Center for -pbc and -center: tric, rect or zero
-box vector 0 0 0 Size for new cubic box (default: read from input)
-clustercenter vector 0 0 0 Optional starting point for pbc cluster option
-trans vector 0 0 0 All coordinates will be translated by trans. This
can advantageously be combined with -pbc mol -ur
compact.
-shift vector 0 0 0 All coordinates will be shifted by framenr*shift
-fit enum none Fit molecule to ref structure in the structure
file: none, rot+trans, rotxy+transxy,
translation, transxy or progressive
-ndec int 3 Precision for .xtc and .gro writing in number of
decimal places
-[no]vel bool yes Read and write velocities if possible
-[no]force bool no Read and write forces if possible
-trunc time -1 Truncate input trajectory file after this time
(ps)
-exec string Execute command for every output frame with the
frame number as argument
-[no]app bool no Append output
-split time 0 Start writing new file when t MOD split = first
time (ps)
-[no]sep bool no Write each frame to a separate .gro, .g96 or .pdb
file
-nzero int 0 If the -sep flag is set, use these many digits
for the file numbers and prepend zeros as needed
-dropunder real 0 Drop all frames below this value
-dropover real 0 Drop all frames above this value
-[no]conect bool no Add conect records when writing .pdb files.
Useful for visualization of non-standard
molecules, e.g. coarse grained ones
Will write pdb: Protein data bank file
-------------------------------------------------------
Program trjconv, VERSION 4.5.5
Source code file: /tmp/build/gromacs-4.5.5/src/gmxlib/gmxfio.c, line: 519
Can not open file:
et_%s.tpr
For more information and tips for troubleshooting, please check the GROMACS
website at http://www.gromacs.org/Documentation/Errors
-------------------------------------------------------
"When It Starts to Start It'll Never Stop" (Magnapop)
--2019-04-21 12:49:51-- http://kodomo.fbb.msu.ru/FBB/year_08/term6/vr.mdp
Resolving kodomo.fbb.msu.ru... 192.168.180.1
Connecting to kodomo.fbb.msu.ru|192.168.180.1|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 1427 (1.4K)
Saving to: `vr.mdp.1'
100%[======================================>] 1,427 --.-K/s in 0s
2019-04-21 12:49:51 (144 MB/s) - `vr.mdp.1' saved [1427/1427]
:-) G R O M A C S (-:
God Rules Over Mankind, Animals, Cosmos and Such
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) grompp (-:
Option Filename Type Description
------------------------------------------------------------
-f vr.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c etane.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p et.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o et_vr.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]v bool no Be loud and noisy
-time real -1 Take frame at or first after this time.
-[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
-maxwarn int 0 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
-[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
-[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Ignoring obsolete mdp entry 'title'
Ignoring obsolete mdp entry 'cpp'
Replacing old mdp entry 'unconstrained-start' by 'continuation'
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.6#
NOTE 1 [file vr.mdp]:
nstcomm < nstcalcenergy defeats the purpose of nstcalcenergy, setting
nstcomm to nstcalcenergy
Generated 332520 of the 332520 non-bonded parameter combinations
Generating 1-4 interactions: fudge = 0.5
Generated 332520 of the 332520 1-4 parameter combinations
Excluding 3 bonded neighbours molecule type 'et'
NOTE 2 [file et.top, line 76]:
System has non-zero total charge: -0.309500
Total charge should normally be an integer. See
http://www.gromacs.org/Documentation/Floating_Point_Arithmetic
for discussion on how close it should be to an integer.
Analysing residue names:
There are: 1 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Number of degrees of freedom in T-Coupling group System is 21.00
NOTE 3 [file vr.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
This run will generate roughly 8 Mb of data
There were 3 notes
Back Off! I just backed up et_vr.tpr to ./#et_vr.tpr.1#
gcq#279: "I Feel a Great Disturbance in the Force" (The Emperor Strikes Back)
:-) G R O M A C S (-:
God Rules Over Mankind, Animals, Cosmos and Such
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s et_vr.tpr Input Run input file: tpr tpb tpa
-o et_vr.trr Output Full precision trajectory: trr trj cpt
-x et_vr.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi et_vr.cpt Input, Opt. Checkpoint file
-cpo et_vr.cpt Output, Opt. Checkpoint file
-c et_vr.gro Output Structure file: gro g96 pdb etc.
-e et_vr.edr Output Energy file
-g et_vr.log Output Log file
-dhdl et_vr.xvg Output, Opt. xvgr/xmgr file
-field et_vr.xvg Output, Opt. xvgr/xmgr file
-table et_vr.xvg Input, Opt. xvgr/xmgr file
-tablep et_vr.xvg Input, Opt. xvgr/xmgr file
-tableb et_vr.xvg Input, Opt. xvgr/xmgr file
-rerun et_vr.trr Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi et_vr.xvg Output, Opt. xvgr/xmgr file
-tpid et_vr.xvg Output, Opt. xvgr/xmgr file
-ei et_vr.edi Input, Opt. ED sampling input
-eo et_vr.edo Output, Opt. ED sampling output
-j et_vr.gct Input, Opt. General coupling stuff
-jo et_vr.gct Output, Opt. General coupling stuff
-ffout et_vr.xvg Output, Opt. xvgr/xmgr file
-devout et_vr.xvg Output, Opt. xvgr/xmgr file
-runav et_vr.xvg Output, Opt. xvgr/xmgr file
-px et_vr.xvg Output, Opt. xvgr/xmgr file
-pf et_vr.xvg Output, Opt. xvgr/xmgr file
-mtx et_vr.mtx Output, Opt. Hessian matrix
-dn et_vr.ndx Output, Opt. Index file
-multidir et_vr Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string et_vr Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 1 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Back Off! I just backed up et_vr.log to ./#et_vr.log.1#
Getting Loaded...
Reading file et_vr.tpr, VERSION 4.5.5 (single precision)
Loaded with Money
Back Off! I just backed up et_vr.trr to ./#et_vr.trr.1#
Back Off! I just backed up et_vr.edr to ./#et_vr.edr.1#
starting mdrun 'first one'
250000 steps, 250.0 ps.
step 249900, remaining runtime: 0 s
Writing final coordinates.
Back Off! I just backed up et_vr.gro to ./#et_vr.gro.1#
step 250000, remaining runtime: 0 s
NODE (s) Real (s) (%)
Time: 3.870 4.156 93.1
(Mnbf/s) (MFlops) (ns/day) (hour/ns)
Performance: 0.000 385.577 5581.418 0.004
gcq#7: "Do the Dog On the Ground" (Red Hot Chili Peppers)
:-) G R O M A C S (-:
Good gRace! Old Maple Actually Chews Slate
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) trjconv (-:
Option Filename Type Description
------------------------------------------------------------
-f et_vr.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-o et_%s.pdb Output Trajectory: xtc trr trj gro g96 pdb
-s et_%s.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-n index.ndx Input, Opt. Index file
-fr frames.ndx Input, Opt. Index file
-sub cluster.ndx Input, Opt. Index file
-drop drop.xvg Input, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-tu enum ps Time unit: fs, ps, ns, us, ms or s
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-skip int 1 Only write every nr-th frame
-dt time 0 Only write frame when t MOD dt = first time (ps)
-[no]round bool no Round measurements to nearest picosecond
-dump time -1 Dump frame nearest specified time (ps)
-t0 time 0 Starting time (ps) (default: don't change)
-timestep time 0 Change time step between input frames (ps)
-pbc enum none PBC treatment (see help text for full
description): none, mol, res, atom, nojump,
cluster or whole
-ur enum rect Unit-cell representation: rect, tric or compact
-[no]center bool no Center atoms in box
-boxcenter enum tric Center for -pbc and -center: tric, rect or zero
-box vector 0 0 0 Size for new cubic box (default: read from input)
-clustercenter vector 0 0 0 Optional starting point for pbc cluster option
-trans vector 0 0 0 All coordinates will be translated by trans. This
can advantageously be combined with -pbc mol -ur
compact.
-shift vector 0 0 0 All coordinates will be shifted by framenr*shift
-fit enum none Fit molecule to ref structure in the structure
file: none, rot+trans, rotxy+transxy,
translation, transxy or progressive
-ndec int 3 Precision for .xtc and .gro writing in number of
decimal places
-[no]vel bool yes Read and write velocities if possible
-[no]force bool no Read and write forces if possible
-trunc time -1 Truncate input trajectory file after this time
(ps)
-exec string Execute command for every output frame with the
frame number as argument
-[no]app bool no Append output
-split time 0 Start writing new file when t MOD split = first
time (ps)
-[no]sep bool no Write each frame to a separate .gro, .g96 or .pdb
file
-nzero int 0 If the -sep flag is set, use these many digits
for the file numbers and prepend zeros as needed
-dropunder real 0 Drop all frames below this value
-dropover real 0 Drop all frames above this value
-[no]conect bool no Add conect records when writing .pdb files.
Useful for visualization of non-standard
molecules, e.g. coarse grained ones
Will write pdb: Protein data bank file
-------------------------------------------------------
Program trjconv, VERSION 4.5.5
Source code file: /tmp/build/gromacs-4.5.5/src/gmxlib/gmxfio.c, line: 519
Can not open file:
et_%s.tpr
For more information and tips for troubleshooting, please check the GROMACS
website at http://www.gromacs.org/Documentation/Errors
-------------------------------------------------------
"Do the Dog On the Ground" (Red Hot Chili Peppers)
--2019-04-21 12:49:56-- http://kodomo.fbb.msu.ru/FBB/year_08/term6/nh.mdp
Resolving kodomo.fbb.msu.ru... 192.168.180.1
Connecting to kodomo.fbb.msu.ru|192.168.180.1|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 1429 (1.4K)
Saving to: `nh.mdp.1'
100%[======================================>] 1,429 --.-K/s in 0s
2019-04-21 12:49:56 (173 MB/s) - `nh.mdp.1' saved [1429/1429]
:-) G R O M A C S (-:
Good gRace! Old Maple Actually Chews Slate
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) grompp (-:
Option Filename Type Description
------------------------------------------------------------
-f nh.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c etane.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p et.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o et_nh.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]v bool no Be loud and noisy
-time real -1 Take frame at or first after this time.
-[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
-maxwarn int 0 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
-[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
-[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Ignoring obsolete mdp entry 'title'
Ignoring obsolete mdp entry 'cpp'
Replacing old mdp entry 'unconstrained-start' by 'continuation'
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.7#
NOTE 1 [file nh.mdp]:
nstcomm < nstcalcenergy defeats the purpose of nstcalcenergy, setting
nstcomm to nstcalcenergy
NOTE 2 [file nh.mdp]:
leapfrog does not yet support Nose-Hoover chains, nhchainlength reset to 1
Generated 332520 of the 332520 non-bonded parameter combinations
Generating 1-4 interactions: fudge = 0.5
Generated 332520 of the 332520 1-4 parameter combinations
Excluding 3 bonded neighbours molecule type 'et'
NOTE 3 [file et.top, line 76]:
System has non-zero total charge: -0.309500
Total charge should normally be an integer. See
http://www.gromacs.org/Documentation/Floating_Point_Arithmetic
for discussion on how close it should be to an integer.
Analysing residue names:
There are: 1 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Number of degrees of freedom in T-Coupling group System is 21.00
NOTE 4 [file nh.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
This run will generate roughly 8 Mb of data
There were 4 notes
Back Off! I just backed up et_nh.tpr to ./#et_nh.tpr.1#
gcq#122: "You Don't Wanna Know" (Pulp Fiction)
:-) G R O M A C S (-:
GROwing Monsters And Cloning Shrimps
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s et_nh.tpr Input Run input file: tpr tpb tpa
-o et_nh.trr Output Full precision trajectory: trr trj cpt
-x et_nh.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi et_nh.cpt Input, Opt. Checkpoint file
-cpo et_nh.cpt Output, Opt. Checkpoint file
-c et_nh.gro Output Structure file: gro g96 pdb etc.
-e et_nh.edr Output Energy file
-g et_nh.log Output Log file
-dhdl et_nh.xvg Output, Opt. xvgr/xmgr file
-field et_nh.xvg Output, Opt. xvgr/xmgr file
-table et_nh.xvg Input, Opt. xvgr/xmgr file
-tablep et_nh.xvg Input, Opt. xvgr/xmgr file
-tableb et_nh.xvg Input, Opt. xvgr/xmgr file
-rerun et_nh.trr Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi et_nh.xvg Output, Opt. xvgr/xmgr file
-tpid et_nh.xvg Output, Opt. xvgr/xmgr file
-ei et_nh.edi Input, Opt. ED sampling input
-eo et_nh.edo Output, Opt. ED sampling output
-j et_nh.gct Input, Opt. General coupling stuff
-jo et_nh.gct Output, Opt. General coupling stuff
-ffout et_nh.xvg Output, Opt. xvgr/xmgr file
-devout et_nh.xvg Output, Opt. xvgr/xmgr file
-runav et_nh.xvg Output, Opt. xvgr/xmgr file
-px et_nh.xvg Output, Opt. xvgr/xmgr file
-pf et_nh.xvg Output, Opt. xvgr/xmgr file
-mtx et_nh.mtx Output, Opt. Hessian matrix
-dn et_nh.ndx Output, Opt. Index file
-multidir et_nh Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string et_nh Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 1 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Back Off! I just backed up et_nh.log to ./#et_nh.log.1#
Getting Loaded...
Reading file et_nh.tpr, VERSION 4.5.5 (single precision)
Loaded with Money
Back Off! I just backed up et_nh.trr to ./#et_nh.trr.1#
Back Off! I just backed up et_nh.edr to ./#et_nh.edr.1#
starting mdrun 'first one'
250000 steps, 250.0 ps.
step 249900, remaining runtime: 0 s
Writing final coordinates.
Back Off! I just backed up et_nh.gro to ./#et_nh.gro.1#
step 250000, remaining runtime: 0 s
NODE (s) Real (s) (%)
Time: 3.890 4.149 93.8
(Mnbf/s) (MFlops) (ns/day) (hour/ns)
Performance: 0.000 383.693 5552.722 0.004
gcq#216: "Everybody is Smashing Things Down" (Offspring)
:-) G R O M A C S (-:
Grunge ROck MAChoS
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) trjconv (-:
Option Filename Type Description
------------------------------------------------------------
-f et_nh.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-o et_%s.pdb Output Trajectory: xtc trr trj gro g96 pdb
-s et_%s.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-n index.ndx Input, Opt. Index file
-fr frames.ndx Input, Opt. Index file
-sub cluster.ndx Input, Opt. Index file
-drop drop.xvg Input, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-tu enum ps Time unit: fs, ps, ns, us, ms or s
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-skip int 1 Only write every nr-th frame
-dt time 0 Only write frame when t MOD dt = first time (ps)
-[no]round bool no Round measurements to nearest picosecond
-dump time -1 Dump frame nearest specified time (ps)
-t0 time 0 Starting time (ps) (default: don't change)
-timestep time 0 Change time step between input frames (ps)
-pbc enum none PBC treatment (see help text for full
description): none, mol, res, atom, nojump,
cluster or whole
-ur enum rect Unit-cell representation: rect, tric or compact
-[no]center bool no Center atoms in box
-boxcenter enum tric Center for -pbc and -center: tric, rect or zero
-box vector 0 0 0 Size for new cubic box (default: read from input)
-clustercenter vector 0 0 0 Optional starting point for pbc cluster option
-trans vector 0 0 0 All coordinates will be translated by trans. This
can advantageously be combined with -pbc mol -ur
compact.
-shift vector 0 0 0 All coordinates will be shifted by framenr*shift
-fit enum none Fit molecule to ref structure in the structure
file: none, rot+trans, rotxy+transxy,
translation, transxy or progressive
-ndec int 3 Precision for .xtc and .gro writing in number of
decimal places
-[no]vel bool yes Read and write velocities if possible
-[no]force bool no Read and write forces if possible
-trunc time -1 Truncate input trajectory file after this time
(ps)
-exec string Execute command for every output frame with the
frame number as argument
-[no]app bool no Append output
-split time 0 Start writing new file when t MOD split = first
time (ps)
-[no]sep bool no Write each frame to a separate .gro, .g96 or .pdb
file
-nzero int 0 If the -sep flag is set, use these many digits
for the file numbers and prepend zeros as needed
-dropunder real 0 Drop all frames below this value
-dropover real 0 Drop all frames above this value
-[no]conect bool no Add conect records when writing .pdb files.
Useful for visualization of non-standard
molecules, e.g. coarse grained ones
Will write pdb: Protein data bank file
-------------------------------------------------------
Program trjconv, VERSION 4.5.5
Source code file: /tmp/build/gromacs-4.5.5/src/gmxlib/gmxfio.c, line: 519
Can not open file:
et_%s.tpr
For more information and tips for troubleshooting, please check the GROMACS
website at http://www.gromacs.org/Documentation/Errors
-------------------------------------------------------
"Everybody is Smashing Things Down" (Offspring)
--2019-04-21 12:50:01-- http://kodomo.fbb.msu.ru/FBB/year_08/term6/an.mdp
Resolving kodomo.fbb.msu.ru... 192.168.180.1
Connecting to kodomo.fbb.msu.ru|192.168.180.1|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 1426 (1.4K)
Saving to: `an.mdp.1'
100%[======================================>] 1,426 --.-K/s in 0s
2019-04-21 12:50:01 (162 MB/s) - `an.mdp.1' saved [1426/1426]
:-) G R O M A C S (-:
Grunge ROck MAChoS
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) grompp (-:
Option Filename Type Description
------------------------------------------------------------
-f an.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c etane.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p et.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o et_an.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]v bool no Be loud and noisy
-time real -1 Take frame at or first after this time.
-[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
-maxwarn int 0 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
-[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
-[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Ignoring obsolete mdp entry 'title'
Ignoring obsolete mdp entry 'cpp'
Replacing old mdp entry 'unconstrained-start' by 'continuation'
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.8#
NOTE 1 [file an.mdp]:
nstcomm < nstcalcenergy defeats the purpose of nstcalcenergy, setting
nstcomm to nstcalcenergy
Generated 332520 of the 332520 non-bonded parameter combinations
Generating 1-4 interactions: fudge = 0.5
Generated 332520 of the 332520 1-4 parameter combinations
Excluding 3 bonded neighbours molecule type 'et'
NOTE 2 [file et.top, line 76]:
System has non-zero total charge: -0.309500
Total charge should normally be an integer. See
http://www.gromacs.org/Documentation/Floating_Point_Arithmetic
for discussion on how close it should be to an integer.
Analysing residue names:
There are: 1 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Number of degrees of freedom in T-Coupling group System is 21.00
NOTE 3 [file an.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
This run will generate roughly 8 Mb of data
There were 3 notes
Back Off! I just backed up et_an.tpr to ./#et_an.tpr.1#
gcq#331: "It's just the way this stuff is done" (Built to Spill)
:-) G R O M A C S (-:
Gravel Rubs Often Many Awfully Cauterized Sores
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s et_an.tpr Input Run input file: tpr tpb tpa
-o et_an.trr Output Full precision trajectory: trr trj cpt
-x et_an.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi et_an.cpt Input, Opt. Checkpoint file
-cpo et_an.cpt Output, Opt. Checkpoint file
-c et_an.gro Output Structure file: gro g96 pdb etc.
-e et_an.edr Output Energy file
-g et_an.log Output Log file
-dhdl et_an.xvg Output, Opt. xvgr/xmgr file
-field et_an.xvg Output, Opt. xvgr/xmgr file
-table et_an.xvg Input, Opt. xvgr/xmgr file
-tablep et_an.xvg Input, Opt. xvgr/xmgr file
-tableb et_an.xvg Input, Opt. xvgr/xmgr file
-rerun et_an.trr Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi et_an.xvg Output, Opt. xvgr/xmgr file
-tpid et_an.xvg Output, Opt. xvgr/xmgr file
-ei et_an.edi Input, Opt. ED sampling input
-eo et_an.edo Output, Opt. ED sampling output
-j et_an.gct Input, Opt. General coupling stuff
-jo et_an.gct Output, Opt. General coupling stuff
-ffout et_an.xvg Output, Opt. xvgr/xmgr file
-devout et_an.xvg Output, Opt. xvgr/xmgr file
-runav et_an.xvg Output, Opt. xvgr/xmgr file
-px et_an.xvg Output, Opt. xvgr/xmgr file
-pf et_an.xvg Output, Opt. xvgr/xmgr file
-mtx et_an.mtx Output, Opt. Hessian matrix
-dn et_an.ndx Output, Opt. Index file
-multidir et_an Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string et_an Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 1 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Back Off! I just backed up et_an.log to ./#et_an.log.1#
Getting Loaded...
Reading file et_an.tpr, VERSION 4.5.5 (single precision)
Loaded with Money
Back Off! I just backed up et_an.trr to ./#et_an.trr.1#
Back Off! I just backed up et_an.edr to ./#et_an.edr.1#
starting mdrun 'first one'
250000 steps, 250.0 ps.
step 249900, remaining runtime: 0 s
Writing final coordinates.
Back Off! I just backed up et_an.gro to ./#et_an.gro.1#
step 250000, remaining runtime: 0 s
NODE (s) Real (s) (%)
Time: 3.790 4.049 93.6
(Mnbf/s) (MFlops) (ns/day) (hour/ns)
Performance: 0.000 393.967 5699.232 0.004
gcq#59: "Move Over Hogey Bear" (Urban Dance Squad)
:-) G R O M A C S (-:
Great Red Owns Many ACres of Sand
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) trjconv (-:
Option Filename Type Description
------------------------------------------------------------
-f et_an.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-o et_%s.pdb Output Trajectory: xtc trr trj gro g96 pdb
-s et_%s.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-n index.ndx Input, Opt. Index file
-fr frames.ndx Input, Opt. Index file
-sub cluster.ndx Input, Opt. Index file
-drop drop.xvg Input, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-tu enum ps Time unit: fs, ps, ns, us, ms or s
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-skip int 1 Only write every nr-th frame
-dt time 0 Only write frame when t MOD dt = first time (ps)
-[no]round bool no Round measurements to nearest picosecond
-dump time -1 Dump frame nearest specified time (ps)
-t0 time 0 Starting time (ps) (default: don't change)
-timestep time 0 Change time step between input frames (ps)
-pbc enum none PBC treatment (see help text for full
description): none, mol, res, atom, nojump,
cluster or whole
-ur enum rect Unit-cell representation: rect, tric or compact
-[no]center bool no Center atoms in box
-boxcenter enum tric Center for -pbc and -center: tric, rect or zero
-box vector 0 0 0 Size for new cubic box (default: read from input)
-clustercenter vector 0 0 0 Optional starting point for pbc cluster option
-trans vector 0 0 0 All coordinates will be translated by trans. This
can advantageously be combined with -pbc mol -ur
compact.
-shift vector 0 0 0 All coordinates will be shifted by framenr*shift
-fit enum none Fit molecule to ref structure in the structure
file: none, rot+trans, rotxy+transxy,
translation, transxy or progressive
-ndec int 3 Precision for .xtc and .gro writing in number of
decimal places
-[no]vel bool yes Read and write velocities if possible
-[no]force bool no Read and write forces if possible
-trunc time -1 Truncate input trajectory file after this time
(ps)
-exec string Execute command for every output frame with the
frame number as argument
-[no]app bool no Append output
-split time 0 Start writing new file when t MOD split = first
time (ps)
-[no]sep bool no Write each frame to a separate .gro, .g96 or .pdb
file
-nzero int 0 If the -sep flag is set, use these many digits
for the file numbers and prepend zeros as needed
-dropunder real 0 Drop all frames below this value
-dropover real 0 Drop all frames above this value
-[no]conect bool no Add conect records when writing .pdb files.
Useful for visualization of non-standard
molecules, e.g. coarse grained ones
Will write pdb: Protein data bank file
-------------------------------------------------------
Program trjconv, VERSION 4.5.5
Source code file: /tmp/build/gromacs-4.5.5/src/gmxlib/gmxfio.c, line: 519
Can not open file:
et_%s.tpr
For more information and tips for troubleshooting, please check the GROMACS
website at http://www.gromacs.org/Documentation/Errors
-------------------------------------------------------
"Move Over Hogey Bear" (Urban Dance Squad)
--2019-04-21 12:50:06-- http://kodomo.fbb.msu.ru/FBB/year_08/term6/sd.mdp
Resolving kodomo.fbb.msu.ru... 192.168.180.1
Connecting to kodomo.fbb.msu.ru|192.168.180.1|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: 1441 (1.4K)
Saving to: `sd.mdp.1'
100%[======================================>] 1,441 --.-K/s in 0s
2019-04-21 12:50:06 (126 MB/s) - `sd.mdp.1' saved [1441/1441]
:-) G R O M A C S (-:
Great Red Owns Many ACres of Sand
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) grompp (-:
Option Filename Type Description
------------------------------------------------------------
-f sd.mdp Input grompp input file with MD parameters
-po mdout.mdp Output grompp input file with MD parameters
-c etane.gro Input Structure file: gro g96 pdb tpr etc.
-r conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-rb conf.gro Input, Opt. Structure file: gro g96 pdb tpr etc.
-n index.ndx Input, Opt. Index file
-p et.top Input Topology file
-pp processed.top Output, Opt. Topology file
-o et_sd.tpr Output Run input file: tpr tpb tpa
-t traj.trr Input, Opt. Full precision trajectory: trr trj cpt
-e ener.edr Input, Opt. Energy file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-[no]v bool no Be loud and noisy
-time real -1 Take frame at or first after this time.
-[no]rmvsbds bool yes Remove constant bonded interactions with virtual
sites
-maxwarn int 0 Number of allowed warnings during input
processing. Not for normal use and may generate
unstable systems
-[no]zero bool no Set parameters for bonded interactions without
defaults to zero instead of generating an error
-[no]renum bool yes Renumber atomtypes and minimize number of
atomtypes
Ignoring obsolete mdp entry 'title'
Ignoring obsolete mdp entry 'cpp'
Replacing old mdp entry 'unconstrained-start' by 'continuation'
Back Off! I just backed up mdout.mdp to ./#mdout.mdp.9#
NOTE 1 [file sd.mdp]:
nstcomm < nstcalcenergy defeats the purpose of nstcalcenergy, setting
nstcomm to nstcalcenergy
Setting the LD random seed to 12983
Generated 332520 of the 332520 non-bonded parameter combinations
Generating 1-4 interactions: fudge = 0.5
Generated 332520 of the 332520 1-4 parameter combinations
Excluding 3 bonded neighbours molecule type 'et'
NOTE 2 [file et.top, line 76]:
System has non-zero total charge: -0.309500
Total charge should normally be an integer. See
http://www.gromacs.org/Documentation/Floating_Point_Arithmetic
for discussion on how close it should be to an integer.
Analysing residue names:
There are: 1 Other residues
Analysing residues not classified as Protein/DNA/RNA/Water and splitting into groups...
Number of degrees of freedom in T-Coupling group System is 21.00
NOTE 3 [file sd.mdp]:
You are using a plain Coulomb cut-off, which might produce artifacts.
You might want to consider using PME electrostatics.
This run will generate roughly 8 Mb of data
There were 3 notes
Back Off! I just backed up et_sd.tpr to ./#et_sd.tpr.1#
gcq#59: "Move Over Hogey Bear" (Urban Dance Squad)
:-) G R O M A C S (-:
S C A M O R G
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s et_sd.tpr Input Run input file: tpr tpb tpa
-o et_sd.trr Output Full precision trajectory: trr trj cpt
-x et_sd.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi et_sd.cpt Input, Opt. Checkpoint file
-cpo et_sd.cpt Output, Opt. Checkpoint file
-c et_sd.gro Output Structure file: gro g96 pdb etc.
-e et_sd.edr Output Energy file
-g et_sd.log Output Log file
-dhdl et_sd.xvg Output, Opt. xvgr/xmgr file
-field et_sd.xvg Output, Opt. xvgr/xmgr file
-table et_sd.xvg Input, Opt. xvgr/xmgr file
-tablep et_sd.xvg Input, Opt. xvgr/xmgr file
-tableb et_sd.xvg Input, Opt. xvgr/xmgr file
-rerun et_sd.trr Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi et_sd.xvg Output, Opt. xvgr/xmgr file
-tpid et_sd.xvg Output, Opt. xvgr/xmgr file
-ei et_sd.edi Input, Opt. ED sampling input
-eo et_sd.edo Output, Opt. ED sampling output
-j et_sd.gct Input, Opt. General coupling stuff
-jo et_sd.gct Output, Opt. General coupling stuff
-ffout et_sd.xvg Output, Opt. xvgr/xmgr file
-devout et_sd.xvg Output, Opt. xvgr/xmgr file
-runav et_sd.xvg Output, Opt. xvgr/xmgr file
-px et_sd.xvg Output, Opt. xvgr/xmgr file
-pf et_sd.xvg Output, Opt. xvgr/xmgr file
-mtx et_sd.mtx Output, Opt. Hessian matrix
-dn et_sd.ndx Output, Opt. Index file
-multidir et_sd Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string et_sd Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 1 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Back Off! I just backed up et_sd.log to ./#et_sd.log.1#
Getting Loaded...
Reading file et_sd.tpr, VERSION 4.5.5 (single precision)
Loaded with Money
Back Off! I just backed up et_sd.trr to ./#et_sd.trr.1#
Back Off! I just backed up et_sd.edr to ./#et_sd.edr.1#
starting mdrun 'first one'
250000 steps, 250.0 ps.
step 249900, remaining runtime: 0 s
Writing final coordinates.
Back Off! I just backed up et_sd.gro to ./#et_sd.gro.1#
step 250000, remaining runtime: 0 s
NODE (s) Real (s) (%)
Time: 4.430 4.710 94.1
(Mnbf/s) (MFlops) (ns/day) (hour/ns)
Performance: 0.000 350.287 4875.866 0.005
gcq#268: "As Always Your Logic Is Impeccable" (Tuvok)
:-) G R O M A C S (-:
God Rules Over Mankind, Animals, Cosmos and Such
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) trjconv (-:
Option Filename Type Description
------------------------------------------------------------
-f et_sd.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-o et_%s.pdb Output Trajectory: xtc trr trj gro g96 pdb
-s et_%s.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-n index.ndx Input, Opt. Index file
-fr frames.ndx Input, Opt. Index file
-sub cluster.ndx Input, Opt. Index file
-drop drop.xvg Input, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-tu enum ps Time unit: fs, ps, ns, us, ms or s
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-skip int 1 Only write every nr-th frame
-dt time 0 Only write frame when t MOD dt = first time (ps)
-[no]round bool no Round measurements to nearest picosecond
-dump time -1 Dump frame nearest specified time (ps)
-t0 time 0 Starting time (ps) (default: don't change)
-timestep time 0 Change time step between input frames (ps)
-pbc enum none PBC treatment (see help text for full
description): none, mol, res, atom, nojump,
cluster or whole
-ur enum rect Unit-cell representation: rect, tric or compact
-[no]center bool no Center atoms in box
-boxcenter enum tric Center for -pbc and -center: tric, rect or zero
-box vector 0 0 0 Size for new cubic box (default: read from input)
-clustercenter vector 0 0 0 Optional starting point for pbc cluster option
-trans vector 0 0 0 All coordinates will be translated by trans. This
can advantageously be combined with -pbc mol -ur
compact.
-shift vector 0 0 0 All coordinates will be shifted by framenr*shift
-fit enum none Fit molecule to ref structure in the structure
file: none, rot+trans, rotxy+transxy,
translation, transxy or progressive
-ndec int 3 Precision for .xtc and .gro writing in number of
decimal places
-[no]vel bool yes Read and write velocities if possible
-[no]force bool no Read and write forces if possible
-trunc time -1 Truncate input trajectory file after this time
(ps)
-exec string Execute command for every output frame with the
frame number as argument
-[no]app bool no Append output
-split time 0 Start writing new file when t MOD split = first
time (ps)
-[no]sep bool no Write each frame to a separate .gro, .g96 or .pdb
file
-nzero int 0 If the -sep flag is set, use these many digits
for the file numbers and prepend zeros as needed
-dropunder real 0 Drop all frames below this value
-dropover real 0 Drop all frames above this value
-[no]conect bool no Add conect records when writing .pdb files.
Useful for visualization of non-standard
molecules, e.g. coarse grained ones
Will write pdb: Protein data bank file
-------------------------------------------------------
Program trjconv, VERSION 4.5.5
Source code file: /tmp/build/gromacs-4.5.5/src/gmxlib/gmxfio.c, line: 519
Can not open file:
et_%s.tpr
For more information and tips for troubleshooting, please check the GROMACS
website at http://www.gromacs.org/Documentation/Errors
-------------------------------------------------------
"As Always Your Logic Is Impeccable" (Tuvok)
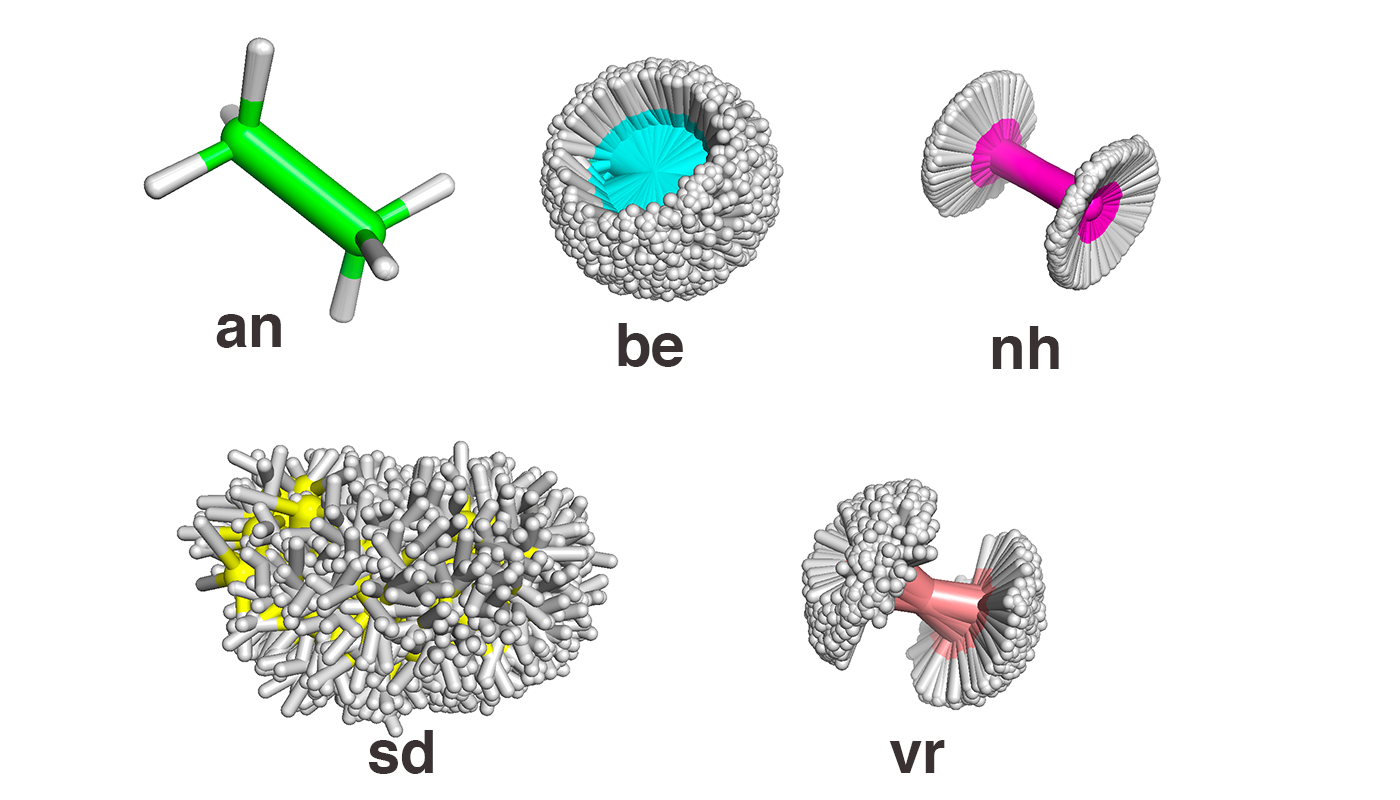
image info
- an (метод Андерсена) - вращения вокруг C-C связи нет
- be (метод Берендсена) - есть вращение вокруг C-C связи + поворот всей молекулы относительно одной оси
- nh (метод Нуза-Хувера) - вращение вокруг C-C связи
- sd (метод стохастической молекулярной динамики) - сильное изменение конформации
- vr (метод "Velocity rescale") - вращение вокруг C-C связи + небольшие отклонения всей молекулы от основной конформации
Сравним потенциальную энергию и кинетическую энергию для каждой из систем.
Изменение потенциальной и кинетической энергии
for i in methods:
!echo 10 11 0 | g_energy -f et_{i}.edr -o et_{i}_en.xvg -xvg none
:-) G R O M A C S (-:
Giant Rising Ordinary Mutants for A Clerical Setup
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_energy (-:
Option Filename Type Description
------------------------------------------------------------
-f et_be.edr Input Energy file
-f2 ener.edr Input, Opt. Energy file
-s topol.tpr Input, Opt. Run input file: tpr tpb tpa
-o et_be_en.xvg Output xvgr/xmgr file
-viol violaver.xvg Output, Opt. xvgr/xmgr file
-pairs pairs.xvg Output, Opt. xvgr/xmgr file
-ora orienta.xvg Output, Opt. xvgr/xmgr file
-ort orientt.xvg Output, Opt. xvgr/xmgr file
-oda orideva.xvg Output, Opt. xvgr/xmgr file
-odr oridevr.xvg Output, Opt. xvgr/xmgr file
-odt oridevt.xvg Output, Opt. xvgr/xmgr file
-oten oriten.xvg Output, Opt. xvgr/xmgr file
-corr enecorr.xvg Output, Opt. xvgr/xmgr file
-vis visco.xvg Output, Opt. xvgr/xmgr file
-ravg runavgdf.xvg Output, Opt. xvgr/xmgr file
-odh dhdl.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-[no]fee bool no Do a free energy estimate
-fetemp real 300 Reference temperature for free energy calculation
-zero real 0 Subtract a zero-point energy
-[no]sum bool no Sum the energy terms selected rather than display
them all
-[no]dp bool no Print energies in high precision
-nbmin int 5 Minimum number of blocks for error estimate
-nbmax int 5 Maximum number of blocks for error estimate
-[no]mutot bool no Compute the total dipole moment from the
components
-skip int 0 Skip number of frames between data points
-[no]aver bool no Also print the exact average and rmsd stored in
the energy frames (only when 1 term is requested)
-nmol int 1 Number of molecules in your sample: the energies
are divided by this number
-[no]driftcorr bool no Useful only for calculations of fluctuation
properties. The drift in the observables will be
subtracted before computing the fluctuation
properties.
-[no]fluc bool no Calculate autocorrelation of energy fluctuations
rather than energy itself
-[no]orinst bool no Analyse instantaneous orientation data
-[no]ovec bool no Also plot the eigenvectors with -oten
-acflen int -1 Length of the ACF, default is half the number of
frames
-[no]normalize bool yes Normalize ACF
-P enum 0 Order of Legendre polynomial for ACF (0 indicates
none): 0, 1, 2 or 3
-fitfn enum none Fit function: none, exp, aexp, exp_exp, vac,
exp5, exp7, exp9 or erffit
-ncskip int 0 Skip this many points in the output file of
correlation functions
-beginfit real 0 Time where to begin the exponential fit of the
correlation function
-endfit real -1 Time where to end the exponential fit of the
correlation function, -1 is until the end
Opened et_be.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Ryckaert-Bell. 4 LJ-14
5 Coulomb-14 6 LJ-(SR) 7 LJ-(LR) 8 Coulomb-(SR)
9 Coulomb-(LR) 10 Potential 11 Kinetic-En. 12 Total-Energy
13 Temperature 14 Pressure 15 Vir-XX 16 Vir-XY
17 Vir-XZ 18 Vir-YX 19 Vir-YY 20 Vir-YZ
21 Vir-ZX 22 Vir-ZY 23 Vir-ZZ 24 Pres-XX
25 Pres-XY 26 Pres-XZ 27 Pres-YX 28 Pres-YY
29 Pres-YZ 30 Pres-ZX 31 Pres-ZY 32 Pres-ZZ
33 #Surf*SurfTen 34 Mu-X 35 Mu-Y 36 Mu-Z
37 T-System 38 Lamb-System
Back Off! I just backed up et_be_en.xvg to ./#et_be_en.xvg.3#
Last energy frame read 2500 time 250.000
Statistics over 250001 steps [ 0.0000 through 250.0000 ps ], 2 data sets
All statistics are over 25001 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Potential 26.3033 0.053 1.68789 0.326111 (kJ/mol)
Kinetic En. 26.1643 0.026 1.69953 0.155248 (kJ/mol)
You may want to use the -driftcorr flag in order to correct
for spurious drift in the graphs. Note that this is not
a substitute for proper equilibration and sampling!
You should select the temperature in order to obtain fluctuation properties.
gcq#184: "You Hear Footsteps Coming From Behind" (Colossal Cave)
:-) G R O M A C S (-:
Giant Rising Ordinary Mutants for A Clerical Setup
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_energy (-:
Option Filename Type Description
------------------------------------------------------------
-f et_vr.edr Input Energy file
-f2 ener.edr Input, Opt. Energy file
-s topol.tpr Input, Opt. Run input file: tpr tpb tpa
-o et_vr_en.xvg Output xvgr/xmgr file
-viol violaver.xvg Output, Opt. xvgr/xmgr file
-pairs pairs.xvg Output, Opt. xvgr/xmgr file
-ora orienta.xvg Output, Opt. xvgr/xmgr file
-ort orientt.xvg Output, Opt. xvgr/xmgr file
-oda orideva.xvg Output, Opt. xvgr/xmgr file
-odr oridevr.xvg Output, Opt. xvgr/xmgr file
-odt oridevt.xvg Output, Opt. xvgr/xmgr file
-oten oriten.xvg Output, Opt. xvgr/xmgr file
-corr enecorr.xvg Output, Opt. xvgr/xmgr file
-vis visco.xvg Output, Opt. xvgr/xmgr file
-ravg runavgdf.xvg Output, Opt. xvgr/xmgr file
-odh dhdl.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-[no]fee bool no Do a free energy estimate
-fetemp real 300 Reference temperature for free energy calculation
-zero real 0 Subtract a zero-point energy
-[no]sum bool no Sum the energy terms selected rather than display
them all
-[no]dp bool no Print energies in high precision
-nbmin int 5 Minimum number of blocks for error estimate
-nbmax int 5 Maximum number of blocks for error estimate
-[no]mutot bool no Compute the total dipole moment from the
components
-skip int 0 Skip number of frames between data points
-[no]aver bool no Also print the exact average and rmsd stored in
the energy frames (only when 1 term is requested)
-nmol int 1 Number of molecules in your sample: the energies
are divided by this number
-[no]driftcorr bool no Useful only for calculations of fluctuation
properties. The drift in the observables will be
subtracted before computing the fluctuation
properties.
-[no]fluc bool no Calculate autocorrelation of energy fluctuations
rather than energy itself
-[no]orinst bool no Analyse instantaneous orientation data
-[no]ovec bool no Also plot the eigenvectors with -oten
-acflen int -1 Length of the ACF, default is half the number of
frames
-[no]normalize bool yes Normalize ACF
-P enum 0 Order of Legendre polynomial for ACF (0 indicates
none): 0, 1, 2 or 3
-fitfn enum none Fit function: none, exp, aexp, exp_exp, vac,
exp5, exp7, exp9 or erffit
-ncskip int 0 Skip this many points in the output file of
correlation functions
-beginfit real 0 Time where to begin the exponential fit of the
correlation function
-endfit real -1 Time where to end the exponential fit of the
correlation function, -1 is until the end
Opened et_vr.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Ryckaert-Bell. 4 LJ-14
5 Coulomb-14 6 LJ-(SR) 7 LJ-(LR) 8 Coulomb-(SR)
9 Coulomb-(LR) 10 Potential 11 Kinetic-En. 12 Total-Energy
13 Conserved-En. 14 Temperature 15 Pressure 16 Vir-XX
17 Vir-XY 18 Vir-XZ 19 Vir-YX 20 Vir-YY
21 Vir-YZ 22 Vir-ZX 23 Vir-ZY 24 Vir-ZZ
25 Pres-XX 26 Pres-XY 27 Pres-XZ 28 Pres-YX
29 Pres-YY 30 Pres-YZ 31 Pres-ZX 32 Pres-ZY
33 Pres-ZZ 34 #Surf*SurfTen 35 Mu-X 36 Mu-Y
37 Mu-Z 38 T-System 39 Lamb-System
Back Off! I just backed up et_vr_en.xvg to ./#et_vr_en.xvg.3#
Last energy frame read 2500 time 250.000
Statistics over 250001 steps [ 0.0000 through 250.0000 ps ], 2 data sets
All statistics are over 25001 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Potential 26.2213 0.23 8.33323 -0.307117 (kJ/mol)
Kinetic En. 25.9963 0.21 7.98625 0.147137 (kJ/mol)
You may want to use the -driftcorr flag in order to correct
for spurious drift in the graphs. Note that this is not
a substitute for proper equilibration and sampling!
You should select the temperature in order to obtain fluctuation properties.
gcq#184: "You Hear Footsteps Coming From Behind" (Colossal Cave)
:-) G R O M A C S (-:
Giant Rising Ordinary Mutants for A Clerical Setup
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_energy (-:
Option Filename Type Description
------------------------------------------------------------
-f et_nh.edr Input Energy file
-f2 ener.edr Input, Opt. Energy file
-s topol.tpr Input, Opt. Run input file: tpr tpb tpa
-o et_nh_en.xvg Output xvgr/xmgr file
-viol violaver.xvg Output, Opt. xvgr/xmgr file
-pairs pairs.xvg Output, Opt. xvgr/xmgr file
-ora orienta.xvg Output, Opt. xvgr/xmgr file
-ort orientt.xvg Output, Opt. xvgr/xmgr file
-oda orideva.xvg Output, Opt. xvgr/xmgr file
-odr oridevr.xvg Output, Opt. xvgr/xmgr file
-odt oridevt.xvg Output, Opt. xvgr/xmgr file
-oten oriten.xvg Output, Opt. xvgr/xmgr file
-corr enecorr.xvg Output, Opt. xvgr/xmgr file
-vis visco.xvg Output, Opt. xvgr/xmgr file
-ravg runavgdf.xvg Output, Opt. xvgr/xmgr file
-odh dhdl.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-[no]fee bool no Do a free energy estimate
-fetemp real 300 Reference temperature for free energy calculation
-zero real 0 Subtract a zero-point energy
-[no]sum bool no Sum the energy terms selected rather than display
them all
-[no]dp bool no Print energies in high precision
-nbmin int 5 Minimum number of blocks for error estimate
-nbmax int 5 Maximum number of blocks for error estimate
-[no]mutot bool no Compute the total dipole moment from the
components
-skip int 0 Skip number of frames between data points
-[no]aver bool no Also print the exact average and rmsd stored in
the energy frames (only when 1 term is requested)
-nmol int 1 Number of molecules in your sample: the energies
are divided by this number
-[no]driftcorr bool no Useful only for calculations of fluctuation
properties. The drift in the observables will be
subtracted before computing the fluctuation
properties.
-[no]fluc bool no Calculate autocorrelation of energy fluctuations
rather than energy itself
-[no]orinst bool no Analyse instantaneous orientation data
-[no]ovec bool no Also plot the eigenvectors with -oten
-acflen int -1 Length of the ACF, default is half the number of
frames
-[no]normalize bool yes Normalize ACF
-P enum 0 Order of Legendre polynomial for ACF (0 indicates
none): 0, 1, 2 or 3
-fitfn enum none Fit function: none, exp, aexp, exp_exp, vac,
exp5, exp7, exp9 or erffit
-ncskip int 0 Skip this many points in the output file of
correlation functions
-beginfit real 0 Time where to begin the exponential fit of the
correlation function
-endfit real -1 Time where to end the exponential fit of the
correlation function, -1 is until the end
Opened et_nh.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Ryckaert-Bell. 4 LJ-14
5 Coulomb-14 6 LJ-(SR) 7 LJ-(LR) 8 Coulomb-(SR)
9 Coulomb-(LR) 10 Potential 11 Kinetic-En. 12 Total-Energy
13 Conserved-En. 14 Temperature 15 Pressure 16 Vir-XX
17 Vir-XY 18 Vir-XZ 19 Vir-YX 20 Vir-YY
21 Vir-YZ 22 Vir-ZX 23 Vir-ZY 24 Vir-ZZ
25 Pres-XX 26 Pres-XY 27 Pres-XZ 28 Pres-YX
29 Pres-YY 30 Pres-YZ 31 Pres-ZX 32 Pres-ZY
33 Pres-ZZ 34 #Surf*SurfTen 35 Mu-X 36 Mu-Y
37 Mu-Z 38 T-System
Back Off! I just backed up et_nh_en.xvg to ./#et_nh_en.xvg.3#
Last energy frame read 2500 time 250.000
Statistics over 250001 steps [ 0.0000 through 250.0000 ps ], 2 data sets
All statistics are over 25001 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Potential 9.23677 0.28 9.37708 1.51803 (kJ/mol)
Kinetic En. 26.0567 0.058 29.1433 0.39633 (kJ/mol)
You may want to use the -driftcorr flag in order to correct
for spurious drift in the graphs. Note that this is not
a substitute for proper equilibration and sampling!
You should select the temperature in order to obtain fluctuation properties.
gcq#184: "You Hear Footsteps Coming From Behind" (Colossal Cave)
:-) G R O M A C S (-:
Giant Rising Ordinary Mutants for A Clerical Setup
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_energy (-:
Option Filename Type Description
------------------------------------------------------------
-f et_an.edr Input Energy file
-f2 ener.edr Input, Opt. Energy file
-s topol.tpr Input, Opt. Run input file: tpr tpb tpa
-o et_an_en.xvg Output xvgr/xmgr file
-viol violaver.xvg Output, Opt. xvgr/xmgr file
-pairs pairs.xvg Output, Opt. xvgr/xmgr file
-ora orienta.xvg Output, Opt. xvgr/xmgr file
-ort orientt.xvg Output, Opt. xvgr/xmgr file
-oda orideva.xvg Output, Opt. xvgr/xmgr file
-odr oridevr.xvg Output, Opt. xvgr/xmgr file
-odt oridevt.xvg Output, Opt. xvgr/xmgr file
-oten oriten.xvg Output, Opt. xvgr/xmgr file
-corr enecorr.xvg Output, Opt. xvgr/xmgr file
-vis visco.xvg Output, Opt. xvgr/xmgr file
-ravg runavgdf.xvg Output, Opt. xvgr/xmgr file
-odh dhdl.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-[no]fee bool no Do a free energy estimate
-fetemp real 300 Reference temperature for free energy calculation
-zero real 0 Subtract a zero-point energy
-[no]sum bool no Sum the energy terms selected rather than display
them all
-[no]dp bool no Print energies in high precision
-nbmin int 5 Minimum number of blocks for error estimate
-nbmax int 5 Maximum number of blocks for error estimate
-[no]mutot bool no Compute the total dipole moment from the
components
-skip int 0 Skip number of frames between data points
-[no]aver bool no Also print the exact average and rmsd stored in
the energy frames (only when 1 term is requested)
-nmol int 1 Number of molecules in your sample: the energies
are divided by this number
-[no]driftcorr bool no Useful only for calculations of fluctuation
properties. The drift in the observables will be
subtracted before computing the fluctuation
properties.
-[no]fluc bool no Calculate autocorrelation of energy fluctuations
rather than energy itself
-[no]orinst bool no Analyse instantaneous orientation data
-[no]ovec bool no Also plot the eigenvectors with -oten
-acflen int -1 Length of the ACF, default is half the number of
frames
-[no]normalize bool yes Normalize ACF
-P enum 0 Order of Legendre polynomial for ACF (0 indicates
none): 0, 1, 2 or 3
-fitfn enum none Fit function: none, exp, aexp, exp_exp, vac,
exp5, exp7, exp9 or erffit
-ncskip int 0 Skip this many points in the output file of
correlation functions
-beginfit real 0 Time where to begin the exponential fit of the
correlation function
-endfit real -1 Time where to end the exponential fit of the
correlation function, -1 is until the end
Opened et_an.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Ryckaert-Bell. 4 LJ-14
5 Coulomb-14 6 LJ-(SR) 7 LJ-(LR) 8 Coulomb-(SR)
9 Coulomb-(LR) 10 Potential 11 Kinetic-En. 12 Total-Energy
13 Temperature 14 Pressure 15 Vir-XX 16 Vir-XY
17 Vir-XZ 18 Vir-YX 19 Vir-YY 20 Vir-YZ
21 Vir-ZX 22 Vir-ZY 23 Vir-ZZ 24 Pres-XX
25 Pres-XY 26 Pres-XZ 27 Pres-YX 28 Pres-YY
29 Pres-YZ 30 Pres-ZX 31 Pres-ZY 32 Pres-ZZ
33 #Surf*SurfTen 34 Mu-X 35 Mu-Y 36 Mu-Z
37 T-System
Back Off! I just backed up et_an_en.xvg to ./#et_an_en.xvg.3#
Last energy frame read 2500 time 250.000
Statistics over 250001 steps [ 0.0000 through 250.0000 ps ], 2 data sets
All statistics are over 25001 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Potential 0.628199 0.00081 0.318084 0.00535395 (kJ/mol)
Kinetic En. 0.759039 0.00086 0.306134 0.00582975 (kJ/mol)
You may want to use the -driftcorr flag in order to correct
for spurious drift in the graphs. Note that this is not
a substitute for proper equilibration and sampling!
You should select the temperature in order to obtain fluctuation properties.
gcq#184: "You Hear Footsteps Coming From Behind" (Colossal Cave)
:-) G R O M A C S (-:
Giant Rising Ordinary Mutants for A Clerical Setup
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_energy (-:
Option Filename Type Description
------------------------------------------------------------
-f et_sd.edr Input Energy file
-f2 ener.edr Input, Opt. Energy file
-s topol.tpr Input, Opt. Run input file: tpr tpb tpa
-o et_sd_en.xvg Output xvgr/xmgr file
-viol violaver.xvg Output, Opt. xvgr/xmgr file
-pairs pairs.xvg Output, Opt. xvgr/xmgr file
-ora orienta.xvg Output, Opt. xvgr/xmgr file
-ort orientt.xvg Output, Opt. xvgr/xmgr file
-oda orideva.xvg Output, Opt. xvgr/xmgr file
-odr oridevr.xvg Output, Opt. xvgr/xmgr file
-odt oridevt.xvg Output, Opt. xvgr/xmgr file
-oten oriten.xvg Output, Opt. xvgr/xmgr file
-corr enecorr.xvg Output, Opt. xvgr/xmgr file
-vis visco.xvg Output, Opt. xvgr/xmgr file
-ravg runavgdf.xvg Output, Opt. xvgr/xmgr file
-odh dhdl.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-[no]fee bool no Do a free energy estimate
-fetemp real 300 Reference temperature for free energy calculation
-zero real 0 Subtract a zero-point energy
-[no]sum bool no Sum the energy terms selected rather than display
them all
-[no]dp bool no Print energies in high precision
-nbmin int 5 Minimum number of blocks for error estimate
-nbmax int 5 Maximum number of blocks for error estimate
-[no]mutot bool no Compute the total dipole moment from the
components
-skip int 0 Skip number of frames between data points
-[no]aver bool no Also print the exact average and rmsd stored in
the energy frames (only when 1 term is requested)
-nmol int 1 Number of molecules in your sample: the energies
are divided by this number
-[no]driftcorr bool no Useful only for calculations of fluctuation
properties. The drift in the observables will be
subtracted before computing the fluctuation
properties.
-[no]fluc bool no Calculate autocorrelation of energy fluctuations
rather than energy itself
-[no]orinst bool no Analyse instantaneous orientation data
-[no]ovec bool no Also plot the eigenvectors with -oten
-acflen int -1 Length of the ACF, default is half the number of
frames
-[no]normalize bool yes Normalize ACF
-P enum 0 Order of Legendre polynomial for ACF (0 indicates
none): 0, 1, 2 or 3
-fitfn enum none Fit function: none, exp, aexp, exp_exp, vac,
exp5, exp7, exp9 or erffit
-ncskip int 0 Skip this many points in the output file of
correlation functions
-beginfit real 0 Time where to begin the exponential fit of the
correlation function
-endfit real -1 Time where to end the exponential fit of the
correlation function, -1 is until the end
Opened et_sd.edr as single precision energy file
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 Bond 2 Angle 3 Ryckaert-Bell. 4 LJ-14
5 Coulomb-14 6 LJ-(SR) 7 LJ-(LR) 8 Coulomb-(SR)
9 Coulomb-(LR) 10 Potential 11 Kinetic-En. 12 Total-Energy
13 Temperature 14 Pressure 15 Vir-XX 16 Vir-XY
17 Vir-XZ 18 Vir-YX 19 Vir-YY 20 Vir-YZ
21 Vir-ZX 22 Vir-ZY 23 Vir-ZZ 24 Pres-XX
25 Pres-XY 26 Pres-XZ 27 Pres-YX 28 Pres-YY
29 Pres-YZ 30 Pres-ZX 31 Pres-ZY 32 Pres-ZZ
33 #Surf*SurfTen 34 Mu-X 35 Mu-Y 36 Mu-Z
37 T-System
Back Off! I just backed up et_sd_en.xvg to ./#et_sd_en.xvg.3#
Last energy frame read 2500 time 250.000
Statistics over 250001 steps [ 0.0000 through 250.0000 ps ], 2 data sets
All statistics are over 25001 points
Energy Average Err.Est. RMSD Tot-Drift
-------------------------------------------------------------------------------
Potential 22.5032 0.24 7.37086 0.897571 (kJ/mol)
Kinetic En. 26.5213 0.25 7.62171 1.04875 (kJ/mol)
You may want to use the -driftcorr flag in order to correct
for spurious drift in the graphs. Note that this is not
a substitute for proper equilibration and sampling!
You should select the temperature in order to obtain fluctuation properties.
gcq#184: "You Hear Footsteps Coming From Behind" (Colossal Cave)
for i, name in zip(methods, ['метод Берендсена', 'метод Velocity rescale',
'метод Нуза-Хувера', 'метод Андерсена', 'метод стохастической молекулярной динамики']):
print(name)
en = np.loadtxt('et_%s_en.xvg' % i)
x = en[:,0]
pot = en[:,1]
kin = en[:,2]
plt.figure(figsize=(12, 8))
plt.plot(x, pot, alpha=0.7, label = 'Potential energy', linewidth=0.5)
plt.plot(x, kin, alpha=0.7, label = 'Kinetic energy', linewidth=0.5)
plt.ylabel('Energy (kJ/mol)')
plt.xlabel('Time (ps)')
plt.legend()
plt.show()
метод Берендсена

метод Velocity rescale

метод Нуза-Хувера

метод Андерсена

метод стохастической молекулярной динамики

Изначально во системах, кроме Андерсена и Нуза-Хувера сравнимые колебания энергии. У Андерсена они слишком маленькие (амплитуда ~ 1.6) и почти не изменяются с течением времени, а у Нуза-Хувера слишком большие (для кинетической ~ 150) и резко уменьшаются спустя 110 ps. В стохастическом метода и в методе Velocity rescale энергия колеблется примерно на одном и том же уровне в течение всего времени. У метода Берендсена же заметно уменьшение амплитуда колебаний уже спустя 20 ps.
Распределение длины связи С-С
for i in methods:
!g_bond -f et_{i}.trr -s et_{i}.tpr -o bond_{i}.xvg -n b.ndx -xvg none
:-) G R O M A C S (-:
GROwing Monsters And Cloning Shrimps
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_bond (-:
Option Filename Type Description
------------------------------------------------------------
-f et_be.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-n b.ndx Input Index file
-s et_be.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-o bond_be.xvg Output xvgr/xmgr file
-l bonds.log Output, Opt. Log file
-d distance.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-dt time 0 Only use frame when t MOD dt = first time (ps)
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-blen real -1 Bond length. By default length of first bond
-tol real 0.1 Half width of distribution as fraction of -blen
-[no]aver bool yes Average bond length distributions
-[no]averdist bool yes Average distances (turns on -d)
Warning: file does not end with a newline, last line:
1 2
Group 0 ( b) has 2 elements
There is one group in the index
Will gather information on 1 bonds
trn version: GMX_trn_file (single precision)
Reading frame 0 time 0.000
Back Off! I just backed up distance.xvg to ./#distance.xvg.5#
Reading frame 200 time 200.000
Total number of samples : 251
Mean : 0.153158
Standard deviation of the distribution: 0.00171314
Standard deviation of the mean : 0.000108132
Back Off! I just backed up bond_be.xvg to ./#bond_be.xvg.1#
gcq#121: "Step On the Brakes" (2 Unlimited)
:-) G R O M A C S (-:
GROwing Monsters And Cloning Shrimps
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_bond (-:
Option Filename Type Description
------------------------------------------------------------
-f et_vr.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-n b.ndx Input Index file
-s et_vr.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-o bond_vr.xvg Output xvgr/xmgr file
-l bonds.log Output, Opt. Log file
-d distance.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-dt time 0 Only use frame when t MOD dt = first time (ps)
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-blen real -1 Bond length. By default length of first bond
-tol real 0.1 Half width of distribution as fraction of -blen
-[no]aver bool yes Average bond length distributions
-[no]averdist bool yes Average distances (turns on -d)
Warning: file does not end with a newline, last line:
1 2
Group 0 ( b) has 2 elements
There is one group in the index
Will gather information on 1 bonds
trn version: GMX_trn_file (single precision)
Reading frame 0 time 0.000
Back Off! I just backed up distance.xvg to ./#distance.xvg.6#
Reading frame 200 time 200.000
Total number of samples : 251
Mean : 0.153312
Standard deviation of the distribution: 0.00329984
Standard deviation of the mean : 0.000208284
Back Off! I just backed up bond_vr.xvg to ./#bond_vr.xvg.1#
gcq#121: "Step On the Brakes" (2 Unlimited)
:-) G R O M A C S (-:
GROwing Monsters And Cloning Shrimps
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_bond (-:
Option Filename Type Description
------------------------------------------------------------
-f et_nh.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-n b.ndx Input Index file
-s et_nh.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-o bond_nh.xvg Output xvgr/xmgr file
-l bonds.log Output, Opt. Log file
-d distance.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-dt time 0 Only use frame when t MOD dt = first time (ps)
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-blen real -1 Bond length. By default length of first bond
-tol real 0.1 Half width of distribution as fraction of -blen
-[no]aver bool yes Average bond length distributions
-[no]averdist bool yes Average distances (turns on -d)
Warning: file does not end with a newline, last line:
1 2
Group 0 ( b) has 2 elements
There is one group in the index
Will gather information on 1 bonds
trn version: GMX_trn_file (single precision)
Reading frame 0 time 0.000
Back Off! I just backed up distance.xvg to ./#distance.xvg.7#
Reading frame 200 time 200.000
Total number of samples : 251
Mean : 0.153245
Standard deviation of the distribution: 0.00298194
Standard deviation of the mean : 0.000188218
Back Off! I just backed up bond_nh.xvg to ./#bond_nh.xvg.1#
gcq#121: "Step On the Brakes" (2 Unlimited)
:-) G R O M A C S (-:
Green Red Orange Magenta Azure Cyan Skyblue
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_bond (-:
Option Filename Type Description
------------------------------------------------------------
-f et_an.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-n b.ndx Input Index file
-s et_an.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-o bond_an.xvg Output xvgr/xmgr file
-l bonds.log Output, Opt. Log file
-d distance.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-dt time 0 Only use frame when t MOD dt = first time (ps)
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-blen real -1 Bond length. By default length of first bond
-tol real 0.1 Half width of distribution as fraction of -blen
-[no]aver bool yes Average bond length distributions
-[no]averdist bool yes Average distances (turns on -d)
Warning: file does not end with a newline, last line:
1 2
Group 0 ( b) has 2 elements
There is one group in the index
Will gather information on 1 bonds
trn version: GMX_trn_file (single precision)
Reading frame 0 time 0.000
Back Off! I just backed up distance.xvg to ./#distance.xvg.8#
Reading frame 200 time 200.000
Total number of samples : 251
Mean : 0.153024
Standard deviation of the distribution: 0.00144019
Standard deviation of the mean : 9.09039e-05
Back Off! I just backed up bond_an.xvg to ./#bond_an.xvg.1#
gcq#236: "Wait a Minute, aren't You.... ? (gunshots) Yeah." (Bodycount)
:-) G R O M A C S (-:
Green Red Orange Magenta Azure Cyan Skyblue
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) g_bond (-:
Option Filename Type Description
------------------------------------------------------------
-f et_sd.trr Input Trajectory: xtc trr trj gro g96 pdb cpt
-n b.ndx Input Index file
-s et_sd.tpr Input, Opt! Structure+mass(db): tpr tpb tpa gro g96 pdb
-o bond_sd.xvg Output xvgr/xmgr file
-l bonds.log Output, Opt. Log file
-d distance.xvg Output, Opt. xvgr/xmgr file
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 19 Set the nicelevel
-b time 0 First frame (ps) to read from trajectory
-e time 0 Last frame (ps) to read from trajectory
-dt time 0 Only use frame when t MOD dt = first time (ps)
-[no]w bool no View output .xvg, .xpm, .eps and .pdb files
-xvg enum none xvg plot formatting: xmgrace, xmgr or none
-blen real -1 Bond length. By default length of first bond
-tol real 0.1 Half width of distribution as fraction of -blen
-[no]aver bool yes Average bond length distributions
-[no]averdist bool yes Average distances (turns on -d)
Warning: file does not end with a newline, last line:
1 2
Group 0 ( b) has 2 elements
There is one group in the index
Will gather information on 1 bonds
trn version: GMX_trn_file (single precision)
Reading frame 0 time 0.000
Back Off! I just backed up distance.xvg to ./#distance.xvg.9#
Reading frame 200 time 200.000
Total number of samples : 251
Mean : 0.153226
Standard deviation of the distribution: 0.00337274
Standard deviation of the mean : 0.000212886
Back Off! I just backed up bond_sd.xvg to ./#bond_sd.xvg.1#
gcq#236: "Wait a Minute, aren't You.... ? (gunshots) Yeah." (Bodycount)
for i, name in zip(methods, ['метод Берендсена', 'метод Velocity rescale',
'метод Нуза-Хувера', 'метод Андерсена', 'метод стохастической молекулярной динамики']):
print(name)
bond = np.loadtxt('bond_%s.xvg' % i)
x = bond[:,0]
y = bond[:,1]
plt.figure(figsize=(12, 8))
plt.bar(x, y, width = 0.0005, label = 'length of C-C bond', color = 'teal', linewidth = 0)
plt.ylabel('Probability density')
plt.xlabel('Bond length (nm)')
plt.legend()
plt.show()
метод Берендсена

метод Velocity rescale

метод Нуза-Хувера

метод Андерсена

метод стохастической молекулярной динамики

У всех методов максимум распределений соответствует длине связи ~0.153. Наименьшая дисперсия у методов Берендсена и Нуза-Хувера .
Распределение потенциальной энергии
for i, name in zip(methods, ['метод Берендсена', 'метод Velocity rescale',
'метод Нуза-Хувера', 'метод Андерсена', 'метод стохастической молекулярной динамики']):
en = np.loadtxt('et_%s_en.xvg' % i)
x = en[:,0]
pot = en[:,1]
print(name)
plt.figure(figsize=(12, 8))
plt.hist(pot, 100, color='teal', linewidth = 0)
plt.show()
метод Берендсена

метод Velocity rescale

метод Нуза-Хувера

метод Андерсена

метод стохастической молекулярной динамики

На распределение Больцмана похоже распределение, полученное методом Нуза-Хувера. Чуть меньше похоже на него распределение, полученное методом Берендсена. Таким образом, эти два метода наиболее реалистично поддерживают температуру в системе.
Быстродействие
Время работы алгоритмов было извлечено из поля Perfomance в .log файлах.
| Method | Perfomance(ns/day) |
|---|---|
| be | 5639.710 |
| vr | 5538.484 |
| nh | 5524.319 |
| an | 5699.232 |
| sd | 4897.979 |
Самый быстрый метод - метод Андерсена, немного уступает ему метод Берендсена. Самый медленный - метод стохастической молекулярной динамики.