Гомологичное моделирование комплекса белка с лигандом
import sys
sys.path.append('/usr/lib/modeller9v7/modlib/')
sys.path.append('/usr/lib/modeller9v7/lib/x86_64-intel8/python2.5/')
import modeller
import _modeller
import modeller.automodel
env=modeller.environ()
env.io.hetatm=True
MODELLER 9v7, 2009/06/12, r6923
PROTEIN STRUCTURE MODELLING BY SATISFACTION OF SPATIAL RESTRAINTS
Copyright(c) 1989-2009 Andrej Sali
All Rights Reserved
Written by A. Sali
with help from
B. Webb, M.S. Madhusudhan, M-Y. Shen, M.A. Marti-Renom,
N. Eswar, F. Alber, M. Topf, B. Oliva, A. Fiser,
R. Sanchez, B. Yerkovich, A. Badretdinov,
F. Melo, J.P. Overington, E. Feyfant
University of California, San Francisco, USA
Rockefeller University, New York, USA
Harvard University, Cambridge, USA
Imperial Cancer Research Fund, London, UK
Birkbeck College, University of London, London, UK
Kind, OS, HostName, Kernel, Processor: 4, Linux kodomo 3.8-2-amd64 x86_64
Date and time of compilation : 2009/06/12 12:23:44
MODELLER executable type : x86_64-intel8
Job starting time (YY/MM/DD HH:MM:SS): 2019/04/25 17:11:04
Моделирование LYS_HYPCU с лигандом
Скачаем белок-заготовку (лизоцим радужной форели):
! wget http://www.rcsb.org/pdb/files/1lmp.pdb
--2019-04-25 17:11:08-- http://www.rcsb.org/pdb/files/1lmp.pdb
Resolving www.rcsb.org... 128.6.244.65
Connecting to www.rcsb.org|128.6.244.65|:80... connected.
HTTP request sent, awaiting response... 301 Moved Permanently
Location: http://files.rcsb.org/view/1lmp.pdb [following]
--2019-04-25 17:11:08-- http://files.rcsb.org/view/1lmp.pdb
Resolving files.rcsb.org... 128.6.244.12
Connecting to files.rcsb.org|128.6.244.12|:80... connected.
HTTP request sent, awaiting response... 200 OK
Length: unspecified [text/plain]
Saving to: `1lmp.pdb.2'
[ <=> ] 130,410 349K/s in 0.4s
2019-04-25 17:11:09 (349 KB/s) - `1lmp.pdb.2' saved [130410]
Скачаем последовательность лизоцима из Hyphantria cunea (LYS_HYPCU):
! wget http://www.uniprot.org/uniprot/P50717.fasta
--2019-04-25 17:11:10-- http://www.uniprot.org/uniprot/P50717.fasta Resolving www.uniprot.org... 193.62.193.81, 128.175.245.185 Connecting to www.uniprot.org|193.62.193.81|:80... connected. HTTP request sent, awaiting response... 301 Moved Permanently Location: https://www.uniprot.org/uniprot/P50717.fasta [following] --2019-04-25 17:11:11-- https://www.uniprot.org/uniprot/P50717.fasta Connecting to www.uniprot.org|193.62.193.81|:443... connected. Unable to establish SSL connection.
Создадим объект выравнивание:
alignm=modeller.alignment(env)
Добавим последовательность и структуру:
alignm.append(file='P50717.fasta', align_codes='all',alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
## есть смысл поправить идентификаторы
alignm[0].code = 'HYPCU'
alignm[1].code = '1LMP'
read_pd_459W> Residue type NAG not recognized. 'automodel' model building
will treat this residue as a rigid body.
To use real parameters, add the residue type to ${LIB}/restyp.lib,
its topology to ${LIB}/top_*.lib, and suitable forcefield
parameters to ${LIB}/par.lib.
read_pd_459W> Residue type NDG not recognized. 'automodel' model building
will treat this residue as a rigid body.
To use real parameters, add the residue type to ${LIB}/restyp.lib,
its topology to ${LIB}/top_*.lib, and suitable forcefield
parameters to ${LIB}/par.lib.
Делаем выравнивание и сохраняем:
alignm.salign()
alignm.write(file='all_in_one.ali', alignment_format='PIR')
SALIGN_____> adding the next group to the alignment; iteration 1
! cat all_in_one.ali
>P1;HYPCU sequence:: : : : :::-1.00:-1.00 MQKLAVFLFAIAAVCIHCEAKYYSTRCDLVRELRKQGFPENQMGDWVCLVENESGRKTDKVGPVNKNGSKDYGLF QINDKYWCSNTRTPGKD--CNVTCADLLLDDITKASTCAKKI-FKRHNFRAWYGWRNHCDGKTLPDTSNC----- -* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELAR------------------ALKASGMDGYAGNSLPNWVCLSKWESSYNTQATN-RNTDGSTDYGIF QINSRYWCDDGRTPGAKNVCGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV.. .*
В последовательности HYPCU нет лиганда. Нужно его добавить. Сделаем это вручную. Результат:
! cat all_with_ligand.ali
>P1;HYPCU sequence:: : : : :::-1.00:-1.00 MQKLAVFLFAIAAVCIHCEAKYYSTRCDLVRELRKQGFPENQMGDWVCLVENESGRKTDKVGPVNKNGSKDYGLF QINDKYWCSNTRTPGKD--CNVTCADLLLDDITKASTCAKKI-FKRHNFRAWYGWRNHCDGKTLPDTSNC---...* >P1;1LMP structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELAR------------------ALKASGMDGYAGNSLPNWVCLSKWESSYNTQATN-RNTDGSTDYGIF QINSRYWCDDGRTPGAKNVCGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV.. .*
Построим модель:
## Выбираем объект для моделирования
s = alignm[0]
pdb = alignm[1]
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='all_with_ligand.ali', knowns= pdb.code, sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 2
a.make()
HYPCU 1LMP
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
92 1 73 76 D C 8.895
END OF TABLE
read_to_681_> topology.submodel read from topology file: 3
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 142
atom names : C +N
atom indices : 1134 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 142
atom names : C CA +N O
atom indices : 1134 1131 0 1135
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 3
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 13318 12314
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_484W> Dihedral still outside +-90: -90.2775
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 145
Number of all, selected real atoms : 1179 1179
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 12314 12314
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2493
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1186.1188
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1161 0 1 0.008 0.008 22.134 1.000
2 Bond angle potential : 1566 1 8 2.345 2.345 171.16 1.000
3 Stereochemical cosine torsion poten: 750 0 30 48.218 48.218 274.82 1.000
4 Stereochemical improper torsion pot: 481 0 0 1.341 1.341 18.318 1.000
5 Soft-sphere overlap restraints : 2493 1 2 0.008 0.008 17.701 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2232 0 3 0.341 0.341 102.78 1.000
10 Distance restraints 2 (N-O) : 2390 0 12 0.478 0.478 182.74 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 141 1 3 5.177 5.177 44.565 1.000
14 Sidechain Chi_1 dihedral restraints: 124 0 3 77.098 77.098 37.280 1.000
15 Sidechain Chi_2 dihedral restraints: 91 0 0 69.198 69.198 39.198 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 70.527 70.527 21.979 1.000
17 Sidechain Chi_4 dihedral restraints: 22 0 0 84.310 84.310 15.701 1.000
18 Disulfide distance restraints : 3 0 0 0.026 0.026 0.33897 1.000
19 Disulfide angle restraints : 6 0 0 2.764 2.764 1.0122 1.000
20 Disulfide dihedral angle restraints: 3 0 0 26.412 26.412 2.0703 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1269 0 0 0.364 0.364 20.704 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 140 20 24 37.669 65.157 160.89 1.000
26 Distance restraints 4 (SDCH-SDCH) : 509 0 0 0.567 0.567 24.981 1.000
27 Distance restraints 5 (X-Y) : 1389 0 2 0.050 0.050 27.747 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: HYPCU.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 16949.4531
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2170 92D 92D N CA 729 730 125.53 107.00 18.54 5.33 107.00 18.54 5.33
-------------------------------------------------------------------------------------------------
Feature 13 : Mainchain Omega dihedral restraints
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 38F 38F CA C 299 307 -155.97 -180.00 24.03 4.80 -180.00 24.03 4.80
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3979 9F 10A C N 75 77 159.91 -134.00 82.27 4.55 -62.50 -164.50 30.19
1 10A 10A N CA 77 78 97.99 147.00 -40.90
2 3980 10A 11I C N 80 82 -73.20 -63.40 10.25 1.72 -120.60 177.36 8.38
2 11I 11I N CA 82 83 -40.61 -43.60 130.30
3 3981 11I 12A C N 88 90 -48.81 -68.20 22.51 1.51 -62.50 175.30 29.42
3 12A 12A N CA 90 91 133.86 145.30 -40.90
4 3982 12A 13A C N 93 95 -166.39 -134.00 35.82 0.81 -62.50 -171.90 36.33
4 13A 13A N CA 95 96 162.30 147.00 -40.90
5 3986 16I 17H C N 119 121 -176.53 -125.60 65.47 1.61 -63.20 178.39 29.36
5 17H 17H N CA 121 122 179.94 138.80 -42.30
6 3988 18C 19E C N 135 137 -134.67 -117.80 27.05 1.47 -63.60 171.39 19.97
6 19E 19E N CA 137 138 115.66 136.80 -40.30
7 3989 19E 20A C N 144 146 -118.39 -134.00 17.64 0.40 -62.50 -171.83 28.67
7 20A 20A N CA 146 147 138.78 147.00 -40.90
8 3990 20A 21K C N 149 151 -82.78 -70.20 69.27 5.30 -62.90 114.82 13.94
8 21K 21K N CA 151 152 72.28 140.40 -40.80
9 3993 23Y 24S C N 182 184 157.49 -136.60 74.74 2.29 -64.10 -164.14 24.09
9 24S 24S N CA 184 185 -173.58 151.20 -35.00
10 3994 24S 25T C N 188 190 -101.49 -124.80 41.92 1.30 -63.20 155.54 17.69
10 25T 25T N CA 190 191 108.66 143.50 -42.10
11 3997 27C 28D C N 212 214 -106.00 -96.50 18.74 0.77 -63.30 144.50 15.76
11 28D 28D N CA 214 215 98.05 114.20 -40.00
12 3999 29L 30V C N 228 230 -159.90 -62.40 97.60 15.68 -62.40 97.60 15.68
12 30V 30V N CA 230 231 -46.71 -42.40 -42.40
13 4023 53E 54S C N 428 430 -137.10 -64.10 79.67 8.54 -64.10 79.67 8.54
13 54S 54S N CA 430 431 -3.08 -35.00 -35.00
14 4054 84S 85N C N 674 676 -117.90 -63.20 72.81 8.30 -63.20 72.81 8.30
14 85N 85N N CA 676 677 6.94 -41.10 -41.10
15 4061 91K 92D C N 727 729 -136.93 -63.30 83.16 14.20 -63.30 83.16 14.20
15 92D 92D N CA 729 730 -78.66 -40.00 -40.00
16 4063 93C 94N C N 741 743 -175.59 -63.20 116.72 18.81 55.90 170.50 10.97
16 94N 94N N CA 743 744 -72.56 -41.10 39.50
17 4085 115I 116F C N 901 903 -44.83 -124.20 92.41 2.44 -63.20 141.46 20.88
17 116F 116F N CA 903 904 95.96 143.30 -44.30
18 4100 130H 131C C N 1050 1052 -114.69 -63.00 52.77 8.17 -63.00 52.77 8.17
18 131C 131C N CA 1052 1053 -30.47 -41.10 -41.10
19 4106 136L 137P C N 1092 1094 -61.69 -64.50 4.49 0.27 -58.70 174.22 14.16
19 137P 137P N CA 1094 1095 143.69 147.20 -30.50
20 4107 137P 138D C N 1099 1101 120.91 -63.30 175.79 27.89 -63.30 175.79 27.89
20 138D 138D N CA 1101 1102 -40.03 -40.00 -40.00
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 8 17 81 85 110 124 133 164 184 182
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 145
Number of all, selected real atoms : 1179 1179
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 12314 12314
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2542
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1211.4454
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1161 0 1 0.009 0.009 24.351 1.000
2 Bond angle potential : 1566 3 12 3.079 3.079 201.90 1.000
3 Stereochemical cosine torsion poten: 750 0 29 48.072 48.072 273.17 1.000
4 Stereochemical improper torsion pot: 481 0 0 1.296 1.296 18.072 1.000
5 Soft-sphere overlap restraints : 2542 1 2 0.008 0.008 17.614 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2232 0 3 0.364 0.364 107.93 1.000
10 Distance restraints 2 (N-O) : 2390 0 11 0.456 0.456 169.83 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 141 1 3 4.801 4.801 38.324 1.000
14 Sidechain Chi_1 dihedral restraints: 124 0 3 77.864 77.864 37.873 1.000
15 Sidechain Chi_2 dihedral restraints: 91 0 0 73.275 73.275 39.114 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 83.940 83.940 27.324 1.000
17 Sidechain Chi_4 dihedral restraints: 22 0 0 71.643 71.643 13.055 1.000
18 Disulfide distance restraints : 3 0 0 0.022 0.022 0.24323 1.000
19 Disulfide angle restraints : 6 0 0 4.130 4.130 2.2592 1.000
20 Disulfide dihedral angle restraints: 3 0 0 41.160 41.160 3.1683 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1269 0 0 0.396 0.396 26.676 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 140 20 23 36.565 66.672 141.43 1.000
26 Distance restraints 4 (SDCH-SDCH) : 509 0 0 0.656 0.656 37.453 1.000
27 Distance restraints 5 (X-Y) : 1389 0 3 0.054 0.054 31.658 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: HYPCU.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 17329.2949
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2170 92D 92D N CA 729 730 127.25 107.00 20.25 5.82 107.00 20.25 5.82
-------------------------------------------------------------------------------------------------
Feature 13 : Mainchain Omega dihedral restraints
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 38F 38F CA C 299 307 -155.56 -180.00 24.44 4.89 -180.00 24.44 4.89
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3979 9F 10A C N 75 77 -90.02 -68.20 55.21 3.71 -62.50 126.12 22.11
1 10A 10A N CA 77 78 -163.98 145.30 -40.90
2 3981 11I 12A C N 88 90 -118.67 -134.00 17.80 0.42 -62.50 -172.55 28.54
2 12A 12A N CA 90 91 137.94 147.00 -40.90
3 3982 12A 13A C N 93 95 -85.01 -68.20 28.16 1.78 -62.50 152.87 26.22
3 13A 13A N CA 95 96 167.90 145.30 -40.90
4 3984 14V 15C C N 105 107 -61.91 -63.00 6.86 0.77 -117.90 179.96 7.77
4 15C 15C N CA 107 108 -47.87 -41.10 141.10
5 3986 16I 17H C N 119 121 -99.42 -125.60 26.25 0.88 -63.20 -177.28 20.83
5 17H 17H N CA 121 122 136.80 138.80 -42.30
6 3988 18C 19E C N 135 137 -60.65 -69.30 9.64 0.58 -63.60 178.58 23.97
6 19E 19E N CA 137 138 138.25 142.50 -40.30
7 3989 19E 20A C N 144 146 -66.18 -68.20 5.75 0.39 -62.50 179.22 29.55
7 20A 20A N CA 146 147 139.92 145.30 -40.90
8 3990 20A 21K C N 149 151 -85.19 -70.20 18.39 1.12 -62.90 169.61 23.31
8 21K 21K N CA 151 152 151.06 140.40 -40.80
9 3993 23Y 24S C N 182 184 91.69 -136.60 133.63 4.74 -64.10 -142.88 17.80
9 24S 24S N CA 184 185 173.77 151.20 -35.00
10 3994 24S 25T C N 188 190 -128.11 -124.80 7.69 0.42 -63.20 -169.92 20.82
10 25T 25T N CA 190 191 136.55 143.50 -42.10
11 3995 25T 26R C N 195 197 -135.40 -125.20 18.38 1.02 -63.00 -178.52 20.81
11 26R 26R N CA 197 198 125.31 140.60 -41.10
12 3997 27C 28D C N 212 214 -123.10 -96.50 40.25 1.65 -63.30 137.66 14.62
12 28D 28D N CA 214 215 84.00 114.20 -40.00
13 3999 29L 30V C N 228 230 -150.82 -62.40 90.40 15.06 -62.40 90.40 15.06
13 30V 30V N CA 230 231 -61.22 -42.40 -42.40
14 4023 53E 54S C N 428 430 -135.79 -64.10 77.78 8.41 -64.10 77.78 8.41
14 54S 54S N CA 430 431 -4.81 -35.00 -35.00
15 4054 84S 85N C N 674 676 -123.71 -63.20 76.77 8.94 -63.20 76.77 8.94
15 85N 85N N CA 676 677 6.14 -41.10 -41.10
16 4061 91K 92D C N 727 729 -177.60 -96.50 90.29 3.68 -63.30 161.79 18.27
16 92D 92D N CA 729 730 74.52 114.20 -40.00
17 4074 104D 105I C N 822 824 -106.99 -63.40 76.61 11.65 -63.40 76.61 11.65
17 105I 105I N CA 824 825 19.40 -43.60 -43.60
18 4085 115I 116F C N 901 903 -47.99 -124.20 89.61 2.35 -63.20 141.30 20.65
18 116F 116F N CA 903 904 96.18 143.30 -44.30
19 4106 136L 137P C N 1092 1094 -62.09 -64.50 9.07 0.57 -58.70 168.99 13.69
19 137P 137P N CA 1094 1095 138.46 147.20 -30.50
20 4107 137P 138D C N 1099 1101 127.62 -63.30 169.08 26.87 -63.30 169.08 26.87
20 138D 138D N CA 1101 1102 -41.02 -40.00 -40.00
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 10 18 81 81 116 121 145 171 211 193
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
HYPCU.B99990001.pdb 1186.11877
HYPCU.B99990002.pdb 1211.44543
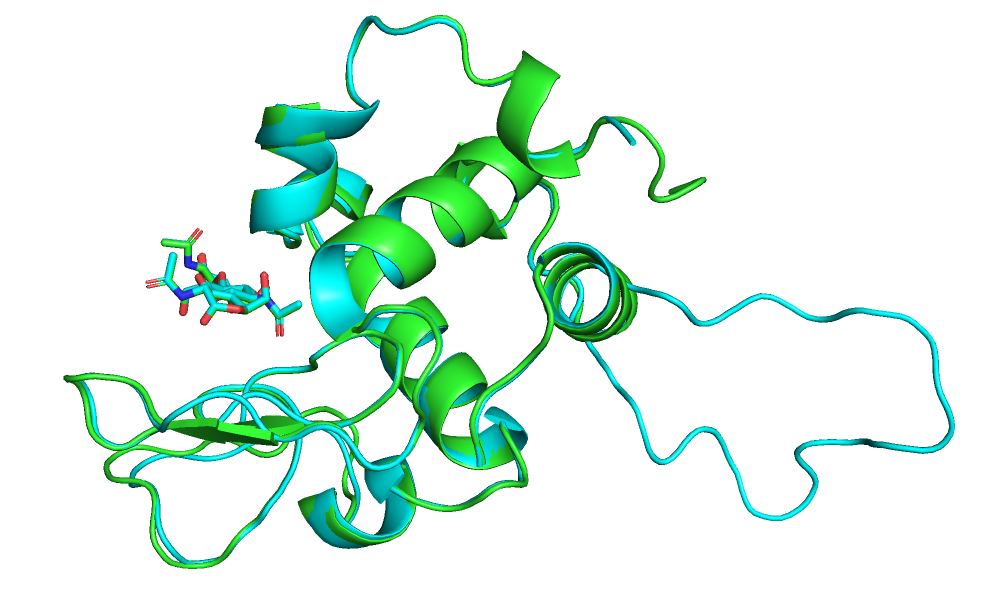
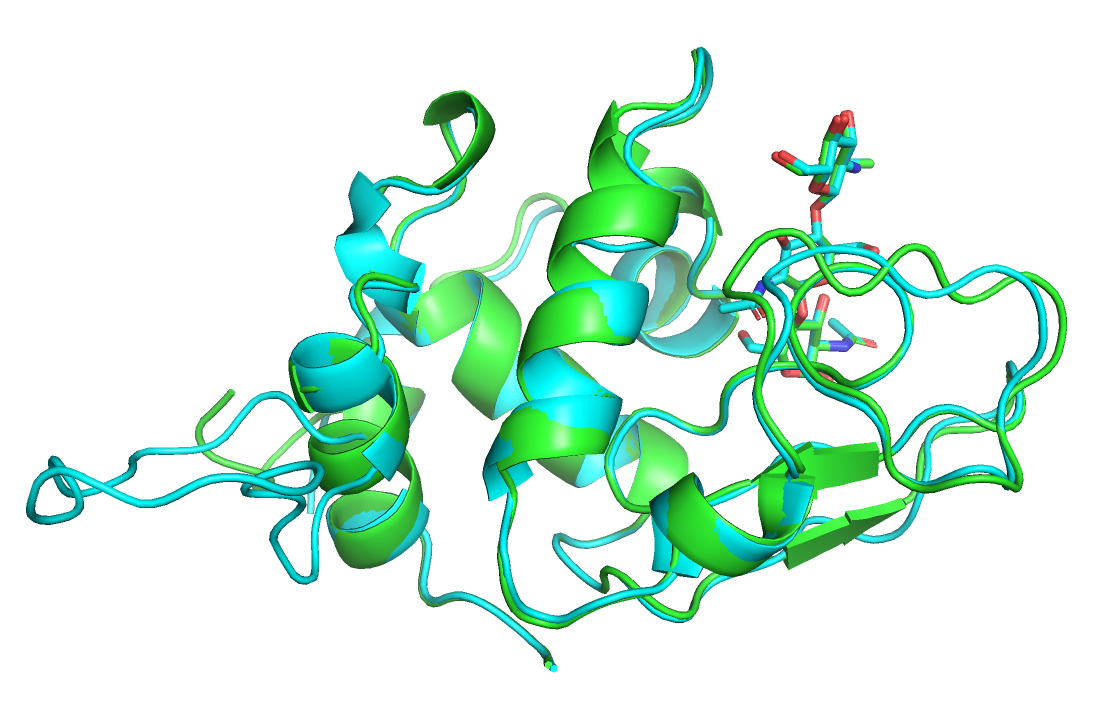
Элементы вторичной структуры выровнены между собой хорошо, отличаютя в основном только петли.
Перемещение лиганда
Попробуем переместить лиганд в другое место.
! rm HYPCU.rsr
class mymodel(modeller.automodel.automodel):
def special_restraints(self, aln):
rsr = self.restraints
at = self.atoms
for x,y in [('O:123','CA:114')]:
rsr.add(modeller.forms.gaussian(group=modeller.physical.xy_distance,
feature=modeller.features.distance(
at[x],at[y]),mean=3.0, stdev=0.1))
from modeller import *
from modeller.automodel import *
a = mymodel(env, alnfile='all_with_ligand.ali', knowns=pdb.code, sequence=s.code)
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
a.name = 'mod' + s.code
a.starting_model = 1
a.ending_model = 2
a.make()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
92 1 73 76 D C 8.895
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 142
atom names : C +N
atom indices : 1134 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 2 142
atom names : C CA +N O
atom indices : 1134 1131 0 1135
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
patch_s_522_> Number of disulfides patched in MODEL: 3
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 13319 12315
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 145
Number of all, selected real atoms : 1179 1179
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 12315 12315
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2500
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1728.8464
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1161 0 1 0.009 0.009 29.679 1.000
2 Bond angle potential : 1566 1 14 2.557 2.557 199.08 1.000
3 Stereochemical cosine torsion poten: 750 0 33 48.912 48.912 280.78 1.000
4 Stereochemical improper torsion pot: 481 1 1 1.533 1.533 28.411 1.000
5 Soft-sphere overlap restraints : 2500 1 2 0.008 0.008 19.644 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2232 7 17 0.526 0.526 244.50 1.000
10 Distance restraints 2 (N-O) : 2390 3 28 0.609 0.609 319.33 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 141 2 6 5.946 5.946 58.794 1.000
14 Sidechain Chi_1 dihedral restraints: 124 0 5 78.192 78.192 50.282 1.000
15 Sidechain Chi_2 dihedral restraints: 91 0 0 75.973 75.973 48.141 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 75.183 75.183 23.404 1.000
17 Sidechain Chi_4 dihedral restraints: 22 0 0 101.766 101.766 15.399 1.000
18 Disulfide distance restraints : 3 0 0 0.026 0.026 0.34163 1.000
19 Disulfide angle restraints : 6 0 0 3.080 3.080 1.2568 1.000
20 Disulfide dihedral angle restraints: 3 0 0 24.324 24.324 1.7401 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1269 0 0 0.457 0.457 33.882 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 140 24 26 39.232 67.638 212.30 1.000
26 Distance restraints 4 (SDCH-SDCH) : 509 0 2 0.898 0.898 55.524 1.000
27 Distance restraints 5 (X-Y) : 1390 2 5 0.078 0.078 106.35 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: HYPCU.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 19570.7070
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 2170 92D 92D N CA 729 730 125.06 107.00 18.06 5.19 107.00 18.06 5.19
-------------------------------------------------------------------------------------------------
Feature 4 : Stereochemical improper torsion potentia
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3895 123A 123A C CA 977 975 13.71 0.00 13.71 4.82 0.00 13.71 4.82
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 8837 123A 115I N O 974 902 9.80 6.71 3.08 5.33 6.71 3.08 5.33
2 8883 125Y 123A N O 993 978 5.37 3.62 1.75 4.91 3.62 1.75 4.91
-------------------------------------------------------------------------------------------------
Feature 13 : Mainchain Omega dihedral restraints
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 38F 38F CA C 299 307 -156.67 -180.00 23.33 4.67 -180.00 23.33 4.67
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3979 9F 10A C N 75 77 -162.41 -134.00 52.99 2.04 -62.50 161.89 31.75
1 10A 10A N CA 77 78 -168.28 147.00 -40.90
2 3981 11I 12A C N 88 90 -117.61 -134.00 25.35 0.84 -62.50 177.34 26.96
2 12A 12A N CA 90 91 127.67 147.00 -40.90
3 3982 12A 13A C N 93 95 -116.33 -134.00 19.48 0.44 -62.50 -172.40 28.63
3 13A 13A N CA 95 96 138.81 147.00 -40.90
4 3988 18C 19E C N 135 137 -144.30 -117.80 44.82 1.59 -63.60 167.48 26.80
4 19E 19E N CA 137 138 172.95 136.80 -40.30
5 3989 19E 20A C N 144 146 -65.94 -68.20 18.89 1.44 -62.50 167.48 27.28
5 20A 20A N CA 146 147 126.55 145.30 -40.90
6 3990 20A 21K C N 149 151 63.37 56.60 27.60 1.53 -62.90 136.80 23.86
6 21K 21K N CA 151 152 11.84 38.60 -40.80
7 3991 21K 22Y C N 158 160 -121.21 -124.30 21.32 1.00 -63.50 167.93 24.03
7 22Y 22Y N CA 160 161 114.30 135.40 -43.40
8 3992 22Y 23Y C N 170 172 -124.27 -124.30 2.40 0.13 -63.50 -173.42 26.82
8 23Y 23Y N CA 172 173 133.00 135.40 -43.40
9 3993 23Y 24S C N 182 184 -109.54 -136.60 35.15 1.09 -64.10 169.95 10.53
9 24S 24S N CA 184 185 128.76 151.20 -35.00
10 3994 24S 25T C N 188 190 -107.25 -124.80 17.78 0.87 -63.20 -177.36 26.36
10 25T 25T N CA 190 191 140.65 143.50 -42.10
11 3995 25T 26R C N 195 197 -139.68 -125.20 14.48 0.57 -63.00 -166.29 31.42
11 26R 26R N CA 197 198 141.02 140.60 -41.10
12 3997 27C 28D C N 212 214 -97.21 -70.90 48.03 1.64 -63.30 133.89 18.71
12 28D 28D N CA 214 215 -169.52 150.30 -40.00
13 3999 29L 30V C N 228 230 -146.84 -62.40 86.13 14.32 -62.40 86.13 14.32
13 30V 30V N CA 230 231 -59.37 -42.40 -42.40
14 4023 53E 54S C N 428 430 -136.35 -64.10 77.86 8.50 -64.10 77.86 8.50
14 54S 54S N CA 430 431 -5.97 -35.00 -35.00
15 4054 84S 85N C N 674 676 -111.52 -119.90 34.60 1.74 -63.20 156.00 22.26
15 85N 85N N CA 676 677 170.58 137.00 -41.10
16 4055 85N 86T C N 682 684 -77.63 -63.20 18.74 2.13 55.90 150.60 18.59
16 86T 86T N CA 684 685 -30.15 -42.10 39.50
17 4061 91K 92D C N 727 729 -142.18 -63.30 85.57 14.57 -63.30 85.57 14.57
17 92D 92D N CA 729 730 -73.17 -40.00 -40.00
18 4074 104D 105I C N 822 824 -110.73 -63.40 79.27 12.06 -63.40 79.27 12.06
18 105I 105I N CA 824 825 19.98 -43.60 -43.60
19 4082 112A 113K C N 875 877 -69.27 -118.00 91.89 3.58 -62.90 102.20 12.94
19 113K 113K N CA 877 878 61.20 139.10 -40.80
20 4083 113K 114K C N 884 886 48.77 -62.90 154.29 18.91 -62.90 154.29 18.91
20 114K 114K N CA 886 887 -147.26 -40.80 -40.80
21 4085 115I 116F C N 901 903 144.96 -124.20 160.11 8.88 -63.20 161.75 22.40
21 116F 116F N CA 903 904 11.46 143.30 -44.30
22 4092 122R 123A C N 972 974 -73.38 -62.50 77.38 12.19 -62.50 77.38 12.19
22 123A 123A N CA 974 975 35.71 -40.90 -40.90
23 4099 129N 130H C N 1040 1042 -121.19 -63.20 80.56 9.13 -63.20 80.56 9.13
23 130H 130H N CA 1042 1043 13.61 -42.30 -42.30
24 4100 130H 131C C N 1050 1052 -126.75 -63.00 63.75 10.60 -63.00 63.75 10.60
24 131C 131C N CA 1052 1053 -40.56 -41.10 -41.10
-------------------------------------------------------------------------------------------------
Feature 27 : Distance restraints 5 (X-Y)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 12315 123A 114K O CA 978 887 4.37 3.00 1.37 13.68 3.00 1.37 13.68
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 10 17 70 79 112 135 145 198 181 186
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
iupac_m_397W> Atoms were not swapped because of the uncertainty of how to handle the H atom.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 145
Number of all, selected real atoms : 1179 1179
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 12315 12315
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 2694
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 2179.2539
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 1161 0 1 0.010 0.010 35.796 1.000
2 Bond angle potential : 1566 4 27 2.963 2.963 270.44 1.000
3 Stereochemical cosine torsion poten: 750 0 32 48.867 48.867 280.21 1.000
4 Stereochemical improper torsion pot: 481 1 3 1.764 1.764 37.485 1.000
5 Soft-sphere overlap restraints : 2694 1 3 0.009 0.009 28.533 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 2232 13 34 0.707 0.707 390.27 1.000
10 Distance restraints 2 (N-O) : 2390 8 52 0.723 0.723 473.18 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 141 4 10 7.727 7.727 99.282 1.000
14 Sidechain Chi_1 dihedral restraints: 124 0 3 70.812 70.812 44.240 1.000
15 Sidechain Chi_2 dihedral restraints: 91 0 1 74.939 74.939 43.784 1.000
16 Sidechain Chi_3 dihedral restraints: 37 0 0 79.360 79.360 23.445 1.000
17 Sidechain Chi_4 dihedral restraints: 22 0 0 72.960 72.960 17.563 1.000
18 Disulfide distance restraints : 3 0 0 0.025 0.025 0.33014 1.000
19 Disulfide angle restraints : 6 0 0 3.809 3.809 1.9221 1.000
20 Disulfide dihedral angle restraints: 3 0 0 22.937 22.937 1.4846 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 1269 0 0 0.493 0.493 41.519 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 140 25 29 37.667 69.473 238.48 1.000
26 Distance restraints 4 (SDCH-SDCH) : 509 0 2 0.821 0.821 41.311 1.000
27 Distance restraints 5 (X-Y) : 1390 1 5 0.078 0.078 109.97 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: HYPCU.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 20309.4805
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1489 30V 31R C N 235 237 140.96 120.00 20.96 4.76 120.00 20.96 4.76
2 2170 92D 92D N CA 729 730 125.80 107.00 18.80 5.40 107.00 18.80 5.40
-------------------------------------------------------------------------------------------------
Feature 4 : Stereochemical improper torsion potentia
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3895 123A 123A C CA 977 975 13.28 0.00 13.28 4.67 0.00 13.28 4.67
-------------------------------------------------------------------------------------------------
Feature 9 : Distance restraints 1 (CA-CA)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4855 29L 105I CA CA 223 825 10.77 8.09 2.68 4.69 8.09 2.68 4.69
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 7036 35K 30V N O 276 236 5.05 2.99 2.06 5.32 2.99 2.06 5.32
2 8837 123A 115I N O 974 902 9.58 6.71 2.86 4.95 6.71 2.86 4.95
3 8860 124W 115I N O 979 902 10.53 7.90 2.63 4.73 7.90 2.63 4.73
4 8883 125Y 123A N O 993 978 5.35 3.62 1.73 4.84 3.62 1.73 4.84
-------------------------------------------------------------------------------------------------
Feature 13 : Mainchain Omega dihedral restraints
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4148 38F 38F CA C 299 307 -155.46 -180.00 24.54 4.91 -180.00 24.54 4.91
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3979 9F 10A C N 75 77 74.83 -68.20 144.25 11.57 -62.50 -143.42 42.64
1 10A 10A N CA 77 78 126.58 145.30 -40.90
2 3981 11I 12A C N 88 90 -92.22 -68.20 49.18 4.69 -62.50 146.33 22.71
2 12A 12A N CA 90 91 102.39 145.30 -40.90
3 3986 16I 17H C N 119 121 -70.37 -67.60 23.85 1.91 -63.20 158.78 19.41
3 17H 17H N CA 121 122 116.31 140.00 -42.30
4 3988 18C 19E C N 135 137 -69.36 -69.30 17.04 1.30 -63.60 160.26 21.68
4 19E 19E N CA 137 138 159.54 142.50 -40.30
5 3989 19E 20A C N 144 146 -51.78 -68.20 16.44 1.37 -62.50 174.94 28.16
5 20A 20A N CA 146 147 144.49 145.30 -40.90
6 3990 20A 21K C N 149 151 -119.28 -118.00 53.15 2.56 -62.90 138.92 21.42
6 21K 21K N CA 151 152 -167.77 139.10 -40.80
7 3992 22Y 23Y C N 170 172 -53.07 -98.40 46.05 1.86 -63.50 -179.79 29.13
7 23Y 23Y N CA 172 173 136.50 128.40 -43.40
8 3993 23Y 24S C N 182 184 -119.68 -136.60 20.30 1.08 -64.10 171.81 16.34
8 24S 24S N CA 184 185 162.42 151.20 -35.00
9 3994 24S 25T C N 188 190 -104.14 -124.80 54.25 1.78 -63.20 141.49 15.80
9 25T 25T N CA 190 191 93.34 143.50 -42.10
10 3995 25T 26R C N 195 197 -112.87 -125.20 13.19 0.42 -63.00 -176.10 21.94
10 26R 26R N CA 197 198 135.91 140.60 -41.10
11 3997 27C 28D C N 212 214 53.21 54.50 10.02 0.84 -63.30 136.42 23.27
11 28D 28D N CA 214 215 30.97 40.90 -40.00
12 4000 30V 31R C N 235 237 -135.69 -63.00 72.71 12.02 -63.00 72.71 12.02
12 31R 31R N CA 237 238 -39.35 -41.10 -41.10
13 4003 33L 34R C N 263 265 159.28 57.30 104.24 11.04 57.30 104.24 11.04
13 34R 34R N CA 265 266 16.40 38.00 38.00
14 4005 35K 36Q C N 283 285 -60.82 -63.80 6.04 1.04 -121.10 -175.15 7.61
14 36Q 36Q N CA 285 286 -35.05 -40.30 139.70
15 4006 36Q 37G C N 292 294 -86.89 -62.40 26.80 4.24 82.20 173.50 12.83
15 37G 37G N CA 294 295 -30.32 -41.20 8.50
16 4054 84S 85N C N 674 676 -110.72 -119.90 32.28 1.65 -63.20 158.26 22.49
16 85N 85N N CA 676 677 167.94 137.00 -41.10
17 4055 85N 86T C N 682 684 -73.16 -63.20 15.44 1.66 55.90 146.72 18.11
17 86T 86T N CA 684 685 -30.30 -42.10 39.50
18 4061 91K 92D C N 727 729 -134.51 -63.30 78.26 13.35 -63.30 78.26 13.35
18 92D 92D N CA 729 730 -72.46 -40.00 -40.00
19 4074 104D 105I C N 822 824 -107.64 -63.40 86.43 13.14 -63.40 86.43 13.14
19 105I 105I N CA 824 825 30.65 -43.60 -43.60
20 4082 112A 113K C N 875 877 -66.73 -118.00 74.79 2.67 -62.90 125.50 16.09
20 113K 113K N CA 877 878 84.65 139.10 -40.80
21 4083 113K 114K C N 884 886 32.81 -62.90 128.29 15.91 -62.90 128.29 15.91
21 114K 114K N CA 886 887 -126.22 -40.80 -40.80
22 4085 115I 116F C N 901 903 50.91 -63.20 167.50 29.83 -63.20 167.50 29.83
22 116F 116F N CA 903 904 78.32 -44.30 -44.30
23 4088 118R 119H C N 932 934 -95.21 -125.60 48.01 1.22 -63.20 147.45 16.68
23 119H 119H N CA 934 935 101.63 138.80 -42.30
24 4092 122R 123A C N 972 974 -74.83 -62.50 81.61 12.82 -62.50 81.61 12.82
24 123A 123A N CA 974 975 39.77 -40.90 -40.90
25 4109 139T 140S C N 1114 1116 -81.69 -64.10 84.45 7.31 -64.10 84.45 7.31
25 140S 140S N CA 1116 1117 -117.60 -35.00 -35.00
-------------------------------------------------------------------------------------------------
Feature 27 : Distance restraints 5 (X-Y)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 12315 123A 114K O CA 978 887 4.44 3.00 1.44 14.36 3.00 1.44 14.36
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 1 14 21 82 91 142 138 156 178 195 183
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
HYPCU.B99990001.pdb 1728.84644
HYPCU.B99990002.pdb 2179.25391
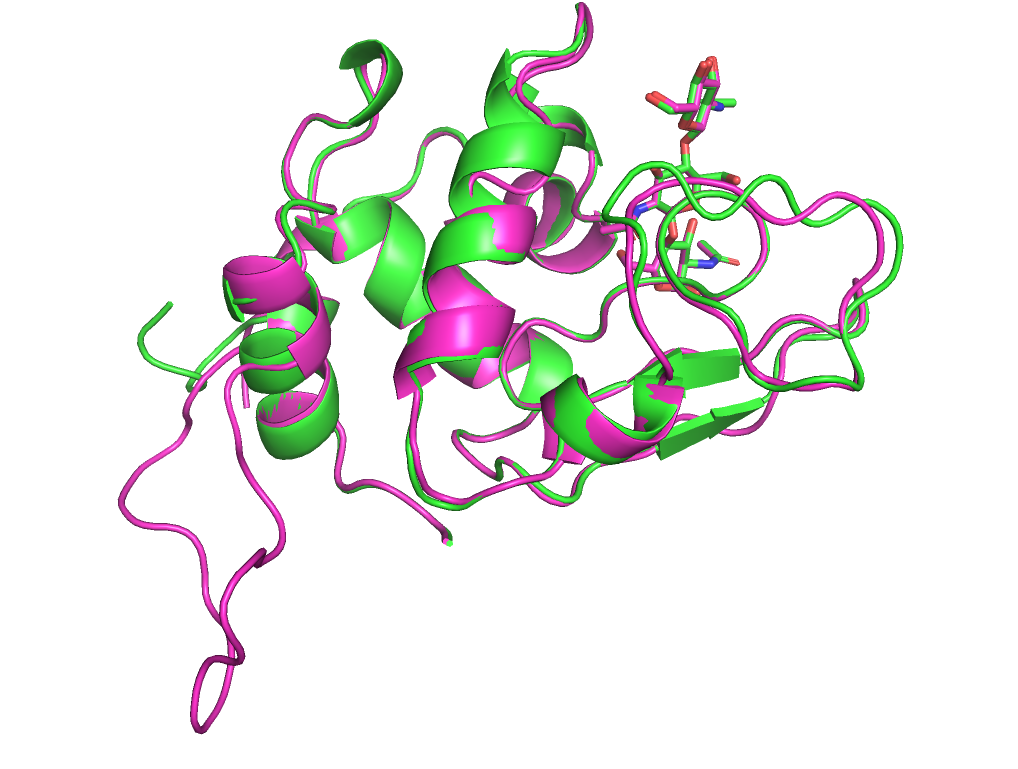
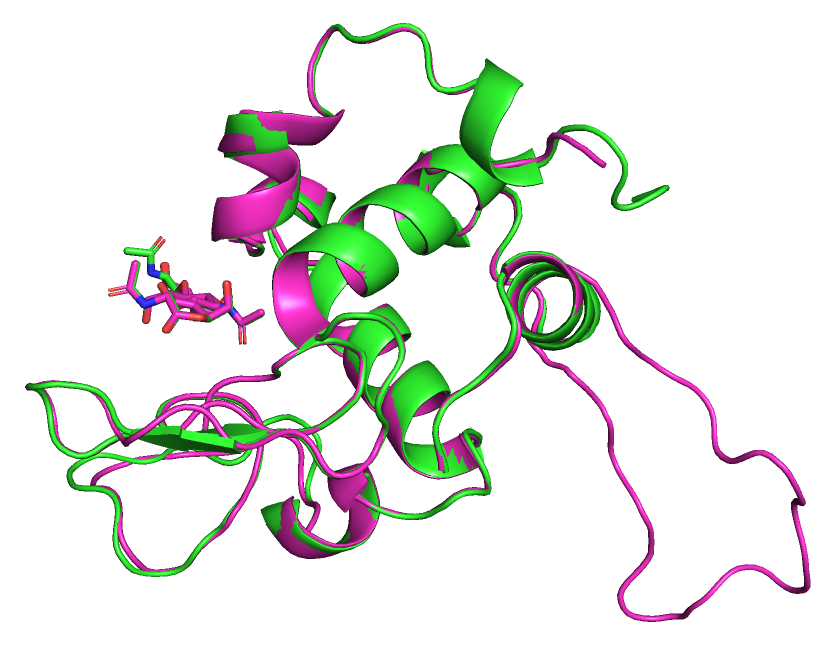
Визуально обе модели так же хорошо выровнены с реферпенсом, как и до перемещения лиганда. Но значения score стали больше.
Замена всех аминокислот на аланин
Сделаем структуру лизоцима с лигандом, заменив все аминокислоты на аланин и сравним score-функцию с "нативной" последовательностью:
alignm = modeller.alignment(env)
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
ala_prot = 'A' * 120 + '...'
alignm.append_sequence(ala_prot)
alignm[0].code = 'pdb'
alignm[1].code = 'ala_prot'
alignm.salign()
alignm.write(file='align_ala.ali', alignment_format='PIR')
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
SALIGN_____> adding the next group to the alignment; iteration 1
! cat align_ala.ali
>P1;pdb structureX:1lmp.pdb: 1 :A:+132 :A:MOL_ID 1; MOLECULE LYSOZYME; CHAIN A; SYNONYM MUCOPEPTIDE N-ACETYLMURAMYLHYDROLASE; EC 3.2.1.17:MOL_ID 1; ORGANISM_SCIENTIFIC ONCORHYNCHUS MYKISS; ORGANISM_COMMON RAINBOW TROUT; ORGANISM_TAXID 8022; ORGAN KIDNEY: 2.00:-1.00 KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGSTDYGIFQINSRYWCDDGRTPGAKNV CGIRCSQLLTDDLTVAIRCAKRVVLDPNGIGAWVAWRLHCQNQDLRSYVAGCGV...* >P1;ala_prot sequence::1 : :+123 : :undefined:undefined:-1.00:-1.00 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA---------AAAAAAAAAAA AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA...*
## Выбираем объект для моделирования
s = alignm[1]
pdb = alignm[0]
print(s.code, pdb.code)
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='align_ala.ali', knowns=pdb.code, sequence=s.code)
a.name = 'mod' + s.code
a.starting_model = 1
a.ending_model = 2
a.make()
('ala_prot', 'pdb')
automodel__W> Topology and/or parameter libraries already in memory. These will
be used instead of the automodel defaults. If this is not what you
want, clear them before creating the automodel object with
env.libs.topology.clear() and env.libs.parameters.clear()
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
check_ali___> Checking the sequence-structure alignment.
Implied target CA(i)-CA(i+1) distances longer than 8.0 angstroms:
ALN_POS TMPL RID1 RID2 NAM1 NAM2 DIST
----------------------------------------------
55 1 55 65 A A 16.730
END OF TABLE
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 1 120
atom names : C +N
atom indices : 599 0
getf_______W> RTF restraint not found in the atoms list:
residue type, indices: 1 120
atom names : C CA +N O
atom indices : 599 597 0 600
fndatmi_285W> Only 129 residues out of 132 contain atoms of type CA
(This is usually caused by non-standard residues, such
as ligands, or by PDB files with missing atoms.)
mdtrsr__446W> A potential that relies on one protein is used, yet you have at
least one known structure available. MDT, not library, potential is used.
iup2crm_280W> No topology library in memory or assigning a BLK residue.
Default CHARMM atom type assigned: C1 --> CT2
This message is written only for the first such atom.
43 atoms in HETATM residues constrained
to protein atoms within 2.30 angstroms
and protein CA atoms within 10.00 angstroms
43 atoms in residues without defined topology
constrained to be rigid bodies
condens_443_> Restraints marked for deletion were removed.
Total number of restraints before, now: 8748 8155
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 123
Number of all, selected real atoms : 644 644
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 8155 8155
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 1132
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1045.0757
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 600 0 1 0.008 0.008 12.573 1.000
2 Bond angle potential : 839 1 4 2.198 2.198 84.385 1.000
3 Stereochemical cosine torsion poten: 244 0 27 73.740 73.740 219.52 1.000
4 Stereochemical improper torsion pot: 240 0 0 0.839 0.839 4.5884 1.000
5 Soft-sphere overlap restraints : 1132 1 2 0.010 0.010 13.093 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 1968 8 31 0.784 0.784 274.76 1.000
10 Distance restraints 2 (N-O) : 2093 7 38 0.764 0.764 361.41 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 119 0 4 4.963 4.963 34.565 1.000
14 Sidechain Chi_1 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
15 Sidechain Chi_2 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
16 Sidechain Chi_3 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
17 Sidechain Chi_4 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
18 Disulfide distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
19 Disulfide angle restraints : 0 0 0 0.000 0.000 0.0000 1.000
20 Disulfide dihedral angle restraints: 0 0 0 0.000 0.000 0.0000 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 668 0 0 0.295 0.295 5.7522 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 118 19 13 27.255 65.368 12.479 1.000
26 Distance restraints 4 (SDCH-SDCH) : 50 0 1 0.972 0.972 3.5846 1.000
27 Distance restraints 5 (X-Y) : 1216 0 0 0.042 0.042 18.366 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: ala_prot.V99990001
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 11267.8242
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 966 53A 53A N CA 261 262 123.87 107.00 16.87 4.85 107.00 16.87 4.85
-------------------------------------------------------------------------------------------------
Feature 9 : Distance restraints 1 (CA-CA)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3583 56A 63A CA CA 277 312 13.39 7.04 6.35 5.99 7.04 6.35 5.99
2 3584 56A 64A CA CA 277 317 13.79 7.69 6.10 4.75 7.69 6.10 4.75
3 3585 56A 65A CA CA 277 322 11.05 5.45 5.60 6.44 5.45 5.60 6.44
4 3588 56A 68A CA CA 277 337 12.22 7.46 4.76 4.62 7.46 4.76 4.62
5 3589 56A 69A CA CA 277 342 9.96 6.07 3.88 5.46 6.07 3.88 5.46
6 3590 56A 70A CA CA 277 347 9.65 4.69 4.96 7.08 4.69 4.96 7.08
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4905 38A 55A N O 186 275 8.24 5.51 2.73 4.66 5.51 2.73 4.66
2 5216 56A 63A N O 276 315 14.14 7.89 6.26 5.08 7.89 6.26 5.08
3 5217 56A 64A N O 276 320 13.33 7.21 6.12 4.65 7.21 6.12 4.65
4 5218 56A 65A N O 276 325 13.69 6.37 7.32 6.92 6.37 7.32 6.92
5 5221 56A 68A N O 276 340 13.11 7.02 6.08 5.68 7.02 6.08 5.68
6 5222 56A 69A N O 276 345 8.84 2.70 6.14 12.00 2.70 6.14 12.00
7 5389 71A 56A N O 351 280 5.67 2.97 2.70 5.70 2.97 2.70 5.70
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1938 15A 16A C N 74 76 79.43 55.40 26.65 2.14 -62.50 157.20 31.94
1 16A 16A N CA 76 77 26.68 38.20 -40.90
2 1940 17A 18A C N 84 86 -56.71 -62.50 7.72 1.21 -134.00 -176.00 11.36
2 18A 18A N CA 86 87 -46.01 -40.90 147.00
3 1943 20A 21A C N 99 101 53.13 55.40 4.12 0.43 -62.50 138.18 28.07
3 21A 21A N CA 101 102 34.76 38.20 -40.90
4 1944 21A 22A C N 104 106 60.90 55.40 11.47 0.42 -62.50 141.39 28.76
4 22A 22A N CA 106 107 28.13 38.20 -40.90
5 1959 36A 37A C N 179 181 67.38 55.40 16.90 0.89 -62.50 146.22 29.73
5 37A 37A N CA 181 182 26.27 38.20 -40.90
6 1960 37A 38A C N 184 186 63.46 55.40 11.76 0.59 -62.50 144.36 29.36
6 38A 38A N CA 186 187 29.64 38.20 -40.90
7 1971 48A 49A C N 239 241 84.54 55.40 39.87 2.21 -62.50 155.93 31.52
7 49A 49A N CA 241 242 10.99 38.20 -40.90
8 1975 52A 53A C N 259 261 -70.26 -62.50 8.64 1.76 -134.00 179.98 11.08
8 53A 53A N CA 261 262 -44.69 -40.90 147.00
9 1976 53A 54A C N 264 266 177.25 -134.00 100.34 4.09 -62.50 146.91 29.80
9 54A 54A N CA 266 267 -125.30 147.00 -40.90
10 1979 56A 57A C N 279 281 -145.36 -134.00 35.98 1.71 -62.50 160.93 30.79
10 57A 57A N CA 281 282 -178.86 147.00 -40.90
11 1980 57A 58A C N 284 286 -71.44 -68.20 22.57 1.70 -62.50 151.73 25.31
11 58A 58A N CA 286 287 167.63 145.30 -40.90
12 1984 61A 62A C N 304 306 66.64 55.40 20.04 0.80 -62.50 143.46 29.16
12 62A 62A N CA 306 307 21.60 38.20 -40.90
13 1986 63A 64A C N 314 316 -81.76 -68.20 14.75 1.04 -62.50 169.10 28.70
13 64A 64A N CA 316 317 151.10 145.30 -40.90
14 1987 64A 65A C N 319 321 -87.79 -68.20 38.01 3.65 -62.50 155.69 24.40
14 65A 65A N CA 321 322 112.73 145.30 -40.90
15 1990 67A 68A C N 334 336 68.34 55.40 20.09 0.93 -62.50 145.53 29.58
15 68A 68A N CA 336 337 22.83 38.20 -40.90
16 2017 94A 95A C N 469 471 80.62 -134.00 159.78 3.63 -62.50 178.00 28.55
16 95A 95A N CA 471 472 -146.73 147.00 -40.90
17 2030 107A 108A C N 534 536 51.35 55.40 6.70 0.29 -62.50 141.74 28.71
17 108A 108A N CA 536 537 43.54 38.20 -40.90
18 2038 115A 116A C N 574 576 59.88 -68.20 142.38 13.74 -68.20 142.38 13.74
18 116A 116A N CA 576 577 -152.51 145.30 145.30
19 2041 118A 119A C N 589 591 50.29 55.40 15.54 0.59 -62.50 146.68 29.59
19 119A 119A N CA 591 592 52.87 38.20 -40.90
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 3 2 35 49 73 51 85 95 98 73
<< end of ENERGY.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NAG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
read_pd_403W> Treating residue type NDG as a BLK (rigid body)
even though topology information appears to be at least
partially available. To treat this residue flexibly
instead, remove the corresponding 'MODELLER BLK RESIDUE'
REMARK from the input PDB file.
>> ENERGY; Differences between the model's features and restraints:
Number of all residues in MODEL : 123
Number of all, selected real atoms : 644 644
Number of all, selected pseudo atoms : 0 0
Number of all static, selected restraints : 8155 8155
COVALENT_CYS : F
NONBONDED_SEL_ATOMS : 1
Number of non-bonded pairs (excluding 1-2,1-3,1-4): 1121
Dynamic pairs routine : 2, NATM x NATM cell sorting
Atomic shift for contacts update (UPDATE_DYNAMIC) : 0.390
LENNARD_JONES_SWITCH : 6.500 7.500
COULOMB_JONES_SWITCH : 6.500 7.500
RESIDUE_SPAN_RANGE : 0 99999
NLOGN_USE : 15
CONTACT_SHELL : 4.000
DYNAMIC_PAIRS,_SPHERE,_COULOMB,_LENNARD,_MODELLER : T T F F F
SPHERE_STDV : 0.050
RADII_FACTOR : 0.820
Current energy : 1058.0981
Summary of the restraint violations:
NUM ... number of restraints.
NUMVI ... number of restraints with RVIOL > VIOL_REPORT_CUT[i].
RVIOL ... relative difference from the best value.
NUMVP ... number of restraints with -Ln(pdf) > VIOL_REPORT_CUT2[i].
RMS_1 ... RMS(feature, minimally_violated_basis_restraint, NUMB).
RMS_2 ... RMS(feature, best_value, NUMB).
MOL.PDF ... scaled contribution to -Ln(Molecular pdf).
# RESTRAINT_GROUP NUM NUMVI NUMVP RMS_1 RMS_2 MOL.PDF S_i
------------------------------------------------------------------------------------------------------
1 Bond length potential : 600 0 1 0.008 0.008 12.865 1.000
2 Bond angle potential : 839 1 3 2.271 2.271 90.685 1.000
3 Stereochemical cosine torsion poten: 244 0 31 74.911 74.911 225.81 1.000
4 Stereochemical improper torsion pot: 240 0 0 0.874 0.874 4.7466 1.000
5 Soft-sphere overlap restraints : 1121 1 2 0.010 0.010 14.064 1.000
6 Lennard-Jones 6-12 potential : 0 0 0 0.000 0.000 0.0000 1.000
7 Coulomb point-point electrostatic p: 0 0 0 0.000 0.000 0.0000 1.000
8 H-bonding potential : 0 0 0 0.000 0.000 0.0000 1.000
9 Distance restraints 1 (CA-CA) : 1968 8 25 0.764 0.764 261.05 1.000
10 Distance restraints 2 (N-O) : 2093 7 34 0.746 0.746 342.49 1.000
11 Mainchain Phi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
12 Mainchain Psi dihedral restraints : 0 0 0 0.000 0.000 0.0000 1.000
13 Mainchain Omega dihedral restraints: 119 0 2 4.832 4.832 32.761 1.000
14 Sidechain Chi_1 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
15 Sidechain Chi_2 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
16 Sidechain Chi_3 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
17 Sidechain Chi_4 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
18 Disulfide distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
19 Disulfide angle restraints : 0 0 0 0.000 0.000 0.0000 1.000
20 Disulfide dihedral angle restraints: 0 0 0 0.000 0.000 0.0000 1.000
21 Lower bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
22 Upper bound distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
23 Distance restraints 3 (SDCH-MNCH) : 668 0 0 0.327 0.327 7.1809 1.000
24 Sidechain Chi_5 dihedral restraints: 0 0 0 0.000 0.000 0.0000 1.000
25 Phi/Psi pair of dihedral restraints: 118 22 18 34.639 74.067 40.167 1.000
26 Distance restraints 4 (SDCH-SDCH) : 50 0 0 1.041 1.041 4.3810 1.000
27 Distance restraints 5 (X-Y) : 1216 0 0 0.047 0.047 21.901 1.000
28 NMR distance restraints 6 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
29 NMR distance restraints 7 (X-Y) : 0 0 0 0.000 0.000 0.0000 1.000
30 Minimal distance restraints : 0 0 0 0.000 0.000 0.0000 1.000
31 Non-bonded restraints : 0 0 0 0.000 0.000 0.0000 1.000
32 Atomic accessibility restraints : 0 0 0 0.000 0.000 0.0000 1.000
33 Atomic density restraints : 0 0 0 0.000 0.000 0.0000 1.000
34 Absolute position restraints : 0 0 0 0.000 0.000 0.0000 1.000
35 Dihedral angle difference restraint: 0 0 0 0.000 0.000 0.0000 1.000
36 GBSA implicit solvent potential : 0 0 0 0.000 0.000 0.0000 1.000
37 EM density fitting potential : 0 0 0 0.000 0.000 0.0000 1.000
38 SAXS restraints : 0 0 0 0.000 0.000 0.0000 1.000
39 Symmetry restraints : 0 0 0 0.000 0.000 0.0000 1.000
# Heavy relative violation of each residue is written to: ala_prot.V99990002
# The profile is NOT normalized by the number of restraints.
# The profiles are smoothed over a window of residues: 1
# The sum of all numbers in the file: 11792.8525
List of the violated restraints:
A restraint is violated when the relative difference
from the best value (RVIOL) is larger than CUTOFF.
ICSR ... index of a restraint in the current set.
RESNO ... residue numbers of the first two atoms.
ATM ... IUPAC atom names of the first two atoms.
FEAT ... the value of the feature in the model.
restr ... the mean of the basis restraint with the smallest
difference from the model (local minimum).
viol ... difference from the local minimum.
rviol ... relative difference from the local minimum.
RESTR ... the best value (global minimum).
VIOL ... difference from the best value.
RVIOL ... relative difference from the best value.
-------------------------------------------------------------------------------------------------
Feature 2 : Bond angle potential
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 910 45A 45A N CA 221 222 124.00 107.00 17.00 4.89 107.00 17.00 4.89
-------------------------------------------------------------------------------------------------
Feature 9 : Distance restraints 1 (CA-CA)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 3583 56A 63A CA CA 277 312 12.15 7.04 5.11 4.82 7.04 5.11 4.82
2 3584 56A 64A CA CA 277 317 13.66 7.69 5.97 4.65 7.69 5.97 4.65
3 3585 56A 65A CA CA 277 322 10.50 5.45 5.05 5.81 5.45 5.05 5.81
4 3588 56A 68A CA CA 277 337 12.17 7.46 4.71 4.57 7.46 4.71 4.57
5 3589 56A 69A CA CA 277 342 10.11 6.07 4.03 5.67 6.07 4.03 5.67
6 3590 56A 70A CA CA 277 347 9.74 4.69 5.05 7.21 4.69 5.05 7.21
-------------------------------------------------------------------------------------------------
Feature 10 : Distance restraints 2 (N-O)
List of the RVIOL violations larger than : 4.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 4905 38A 55A N O 186 275 8.24 5.51 2.73 4.66 5.51 2.73 4.66
2 5216 56A 63A N O 276 315 14.33 7.89 6.44 5.23 7.89 6.44 5.23
3 5217 56A 64A N O 276 320 14.42 7.21 7.20 5.47 7.21 7.20 5.47
4 5218 56A 65A N O 276 325 12.90 6.37 6.54 6.18 6.37 6.54 6.18
5 5221 56A 68A N O 276 340 12.40 7.02 5.38 5.02 7.02 5.38 5.02
6 5222 56A 69A N O 276 345 9.04 2.70 6.34 12.39 2.70 6.34 12.39
7 5389 71A 56A N O 351 280 5.74 2.97 2.76 5.84 2.97 2.76 5.84
-------------------------------------------------------------------------------------------------
Feature 25 : Phi/Psi pair of dihedral restraints
List of the RVIOL violations larger than : 6.5000
# ICSR RESNO1/2 ATM1/2 INDATM1/2 FEAT restr viol rviol RESTR VIOL RVIOL
1 1938 15A 16A C N 74 76 79.23 55.40 27.38 2.05 -62.50 156.18 31.73
1 16A 16A N CA 76 77 24.72 38.20 -40.90
2 1940 17A 18A C N 84 86 -51.89 -62.50 10.61 2.03 -134.00 -169.48 11.76
2 18A 18A N CA 86 87 -41.08 -40.90 147.00
3 1941 18A 19A C N 89 91 -137.83 -134.00 76.07 4.40 -62.50 134.91 20.28
3 19A 19A N CA 91 92 71.03 147.00 -40.90
4 1943 20A 21A C N 99 101 55.80 55.40 3.67 0.19 -62.50 140.31 28.51
4 21A 21A N CA 101 102 34.55 38.20 -40.90
5 1944 21A 22A C N 104 106 62.66 55.40 10.36 0.54 -62.50 144.24 29.33
5 22A 22A N CA 106 107 30.81 38.20 -40.90
6 1959 36A 37A C N 179 181 66.87 55.40 14.72 0.90 -62.50 147.03 29.90
6 37A 37A N CA 181 182 28.97 38.20 -40.90
7 1960 37A 38A C N 184 186 60.83 55.40 9.64 0.39 -62.50 142.37 28.95
7 38A 38A N CA 186 187 30.23 38.20 -40.90
8 1967 44A 45A C N 219 221 -71.25 -62.50 12.79 2.54 -68.20 164.50 13.16
8 45A 45A N CA 221 222 -50.23 -40.90 145.30
9 1971 48A 49A C N 239 241 162.86 -134.00 65.94 1.59 -62.50 -156.11 40.37
9 49A 49A N CA 241 242 166.00 147.00 -40.90
10 1972 49A 50A C N 244 246 81.53 -134.00 144.55 4.61 -68.20 149.87 12.92
10 50A 50A N CA 246 247 151.81 147.00 145.30
11 1976 53A 54A C N 264 266 59.16 -68.20 142.63 13.80 -62.50 163.74 25.61
11 54A 54A N CA 266 267 -150.48 145.30 -40.90
12 1979 56A 57A C N 279 281 -152.62 -134.00 39.62 1.64 -62.50 164.10 31.67
12 57A 57A N CA 281 282 -178.04 147.00 -40.90
13 1980 57A 58A C N 284 286 -73.09 -68.20 19.47 1.40 -62.50 155.31 25.99
13 58A 58A N CA 286 287 164.15 145.30 -40.90
14 1983 60A 61A C N 299 301 -60.84 -62.50 1.89 0.38 -68.20 174.85 14.36
14 61A 61A N CA 301 302 -40.00 -40.90 145.30
15 1985 62A 63A C N 309 311 47.86 55.40 30.58 1.28 -134.00 -165.06 9.51
15 63A 63A N CA 311 312 67.84 38.20 147.00
16 1987 64A 65A C N 319 321 49.89 55.40 35.45 1.70 -62.50 160.16 31.96
16 65A 65A N CA 321 322 73.22 38.20 -40.90
17 1989 66A 67A C N 329 331 -81.47 -68.20 43.01 3.91 -62.50 146.52 23.15
17 67A 67A N CA 331 332 104.39 145.30 -40.90
18 1990 67A 68A C N 334 336 -61.90 -68.20 37.66 2.81 -62.50 149.07 24.46
18 68A 68A N CA 336 337 108.17 145.30 -40.90
19 2017 94A 95A C N 469 471 79.67 -134.00 161.10 3.66 -62.50 176.57 28.34
19 95A 95A N CA 471 472 -145.62 147.00 -40.90
20 2030 107A 108A C N 534 536 49.52 55.40 9.92 0.42 -62.50 141.88 28.70
20 108A 108A N CA 536 537 46.19 38.20 -40.90
21 2039 116A 117A C N 579 581 68.93 55.40 40.35 1.52 -62.50 137.70 27.75
21 117A 117A N CA 581 582 0.19 38.20 -40.90
22 2041 118A 119A C N 589 591 134.47 -134.00 128.24 3.82 -62.50 -177.38 37.13
22 119A 119A N CA 591 592 -123.18 147.00 -40.90
report______> Distribution of short non-bonded contacts:
DISTANCE1: 0.00 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40
DISTANCE2: 2.10 2.20 2.30 2.40 2.50 2.60 2.70 2.80 2.90 3.00 3.10 3.20 3.30 3.40 3.50
FREQUENCY: 0 0 0 0 0 4 4 45 42 72 46 85 90 95 73
<< end of ENERGY.
>> Summary of successfully produced models:
Filename molpdf
----------------------------------------
ala_prot.B99990001.pdb 1045.07568
ala_prot.B99990002.pdb 1058.09814
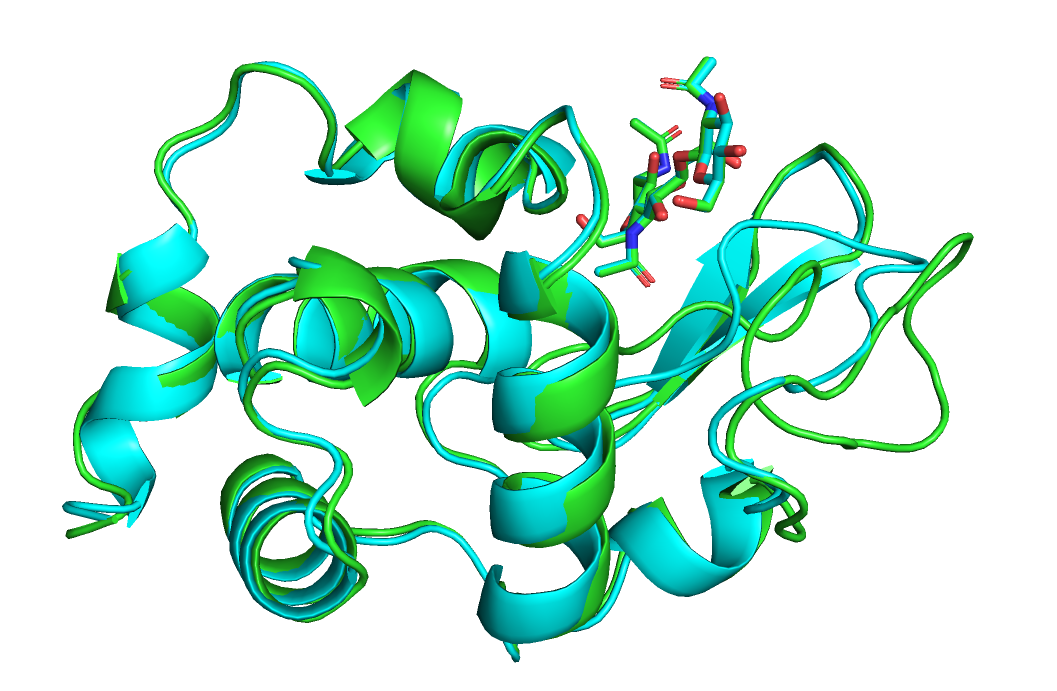
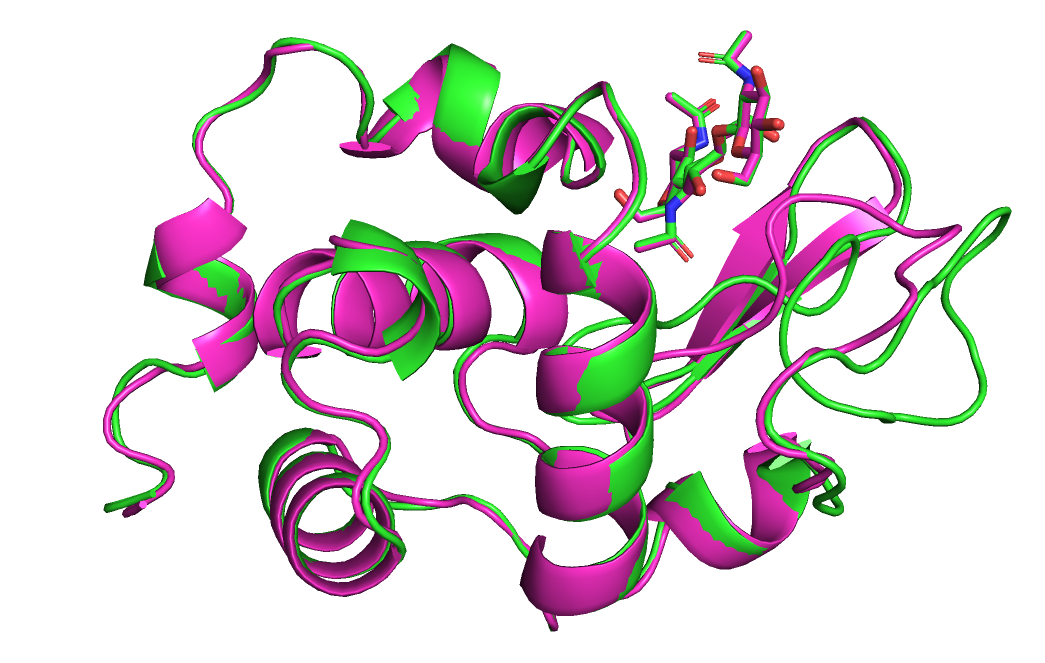
Модель, состоящую только из аланинов тоже можно свернуть как референс (лизоцим радуной форели, 1LMP) - элементы вторичной структуры достаточно хорошо выравниваются, основные различия наблюдаются в петлях. Причем scores, которые при этом получаются, сопоставимы с тем, что были получены при моделировании гомологичного белка.