Практикум 8. Поиск по сходству (BLAST)
Task 1
В рамках задания нужно было проаннотировать последовательность ДНК, полученную в 6 практикуме. Алгоритм megablast выводит наиболее похожие на введенную последовательности (в смысле выравнивания), что позволяет в большинстве случаев найти наиболее близких гомологов введенной последовательности, что говорит об эволюционной близости последовательностей, а в случае ортологов - о близком родстве организмов-носителей последовательностей. Поэтому для идентификации видовой принадлежности нукдеотидной последовательности разумно было воспользоваться этим алгоритмом (в разделе blastn).
Параметры поиска брались по умолчанию. Алгоритм вывел на экран 100 последовательностей, все из них относятся к митохондриальному гену субъединицы 1 белка цитохромоксидазы (CO1), одного из генов, широко использующегося в баркодировании. 99 из 100 выведенных последовательностей относились к видам офиур, причем для всех E-value меньше e-145 последовательности принадлежали роду Ophiopholis, а при проценте Identity больше 90 последовательности принадлежали конкретному виду O. aculeata - самому распространенному на Белом море виду офиур. Нонсенсом и досадной ошибкой показалась нам выдача последовательности этого же митохондриального гена, принадлежащего краснокнижной бабочке Agriades zullichi, эндемичной для Испании: несмотря на очень хорошее качество выравнивания (E-value ~ 0, identity 95.78%), очевидно, что гены очень давно разошлись эволюционно, как и организмы. Вероятно, это связано с ошибками базы данных.
Вот наиболее яркие последовательности, выданные алгоритмом:
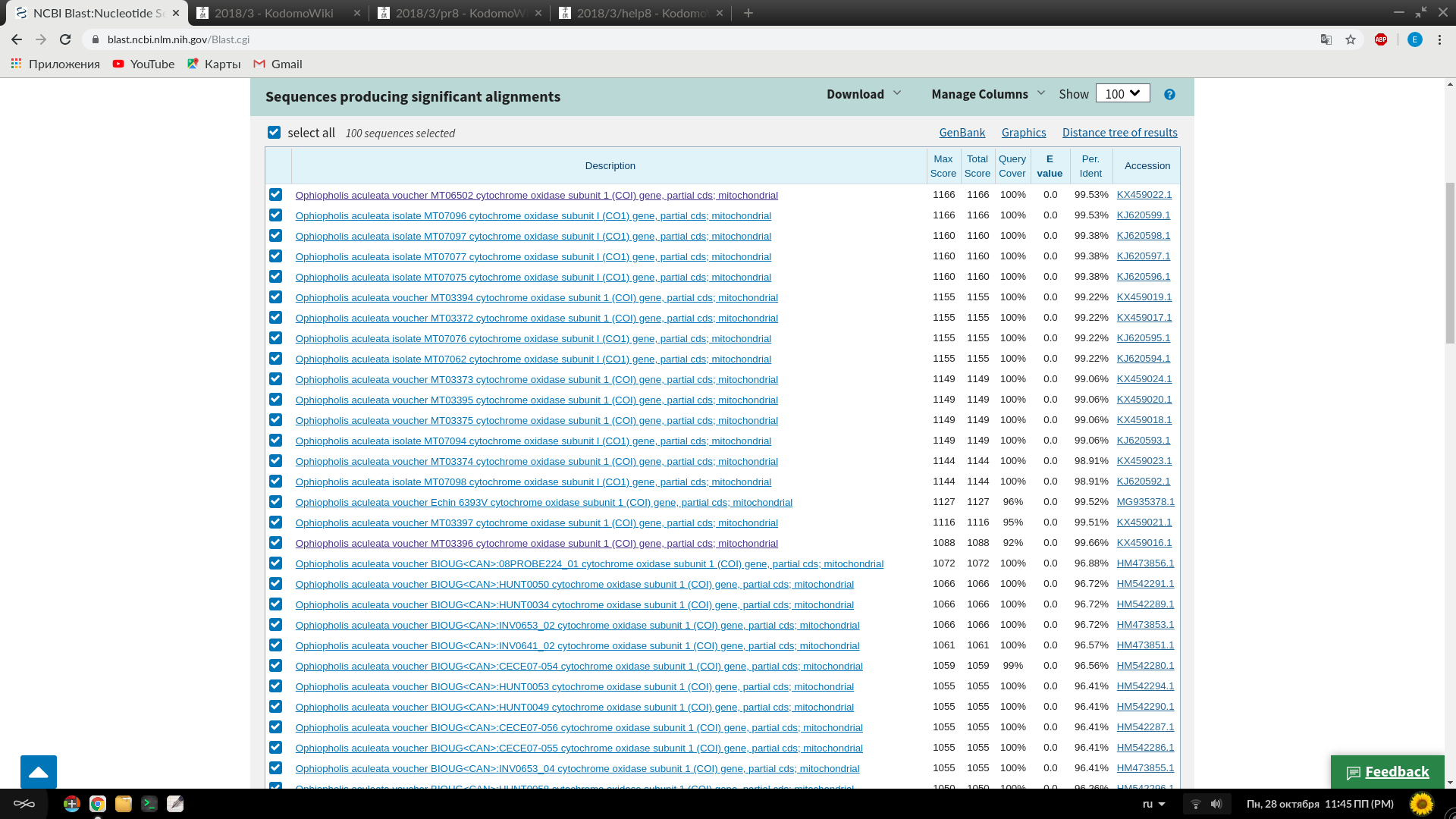
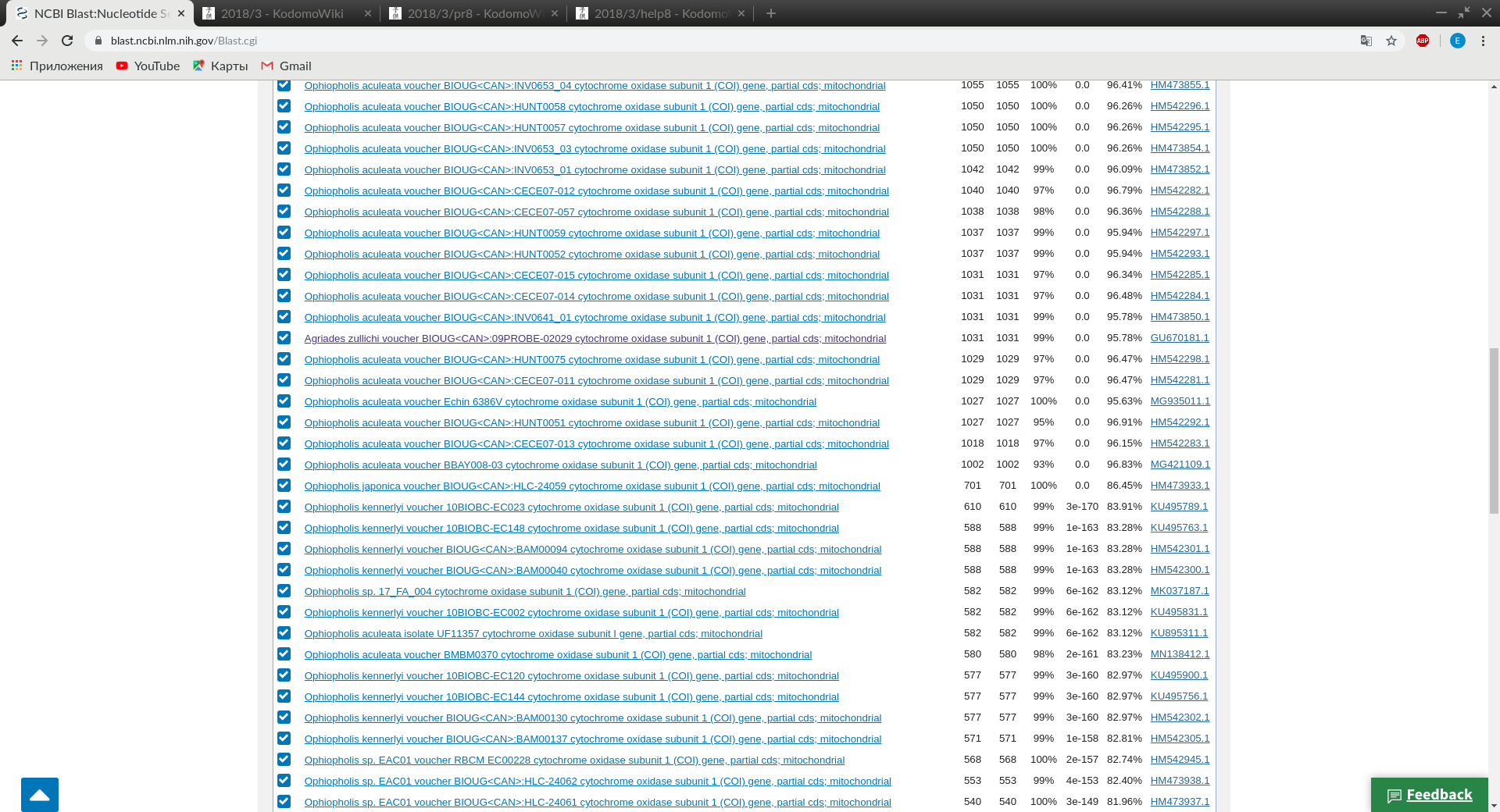
На основе работы алгоритма можно проаннотировать последовательность:

| Latin name | Ophiopholis aculeata |
|---|---|
| Taxonomy | ***look higher*** |
| Gene | cytochrome oxidase subunit 1 (COI) gene, partial cds; mitochondrial |
| Minimal reliable Identity | 95.78 |
| Maximal reliable Evalue | 0 |
| Minimal reliable Score | 1002 |
Task 2
В рамках задания требовалось сравнить результаты работы алгоритмов выравнивания с разными параметрами со входными последовательностями из 6 и 7 (вирусный CDS) практикумов. Предложено было 3 варианта работы алгоритма: megablast, blastn (параметры по умолчанию), blastn (оптимальные параметры).
Результаты работы с последовательностью CO1 были достаточно ск учны: в базе данных много последовательностей, слишком хорошо выравнивающихся с ней. Поэтому различия при использовании алгоритмов были несущественны. Оптимальные параметры - длина слова 7 и ограничение E-value 10^-3 - не сидьгно повлияли на результат.

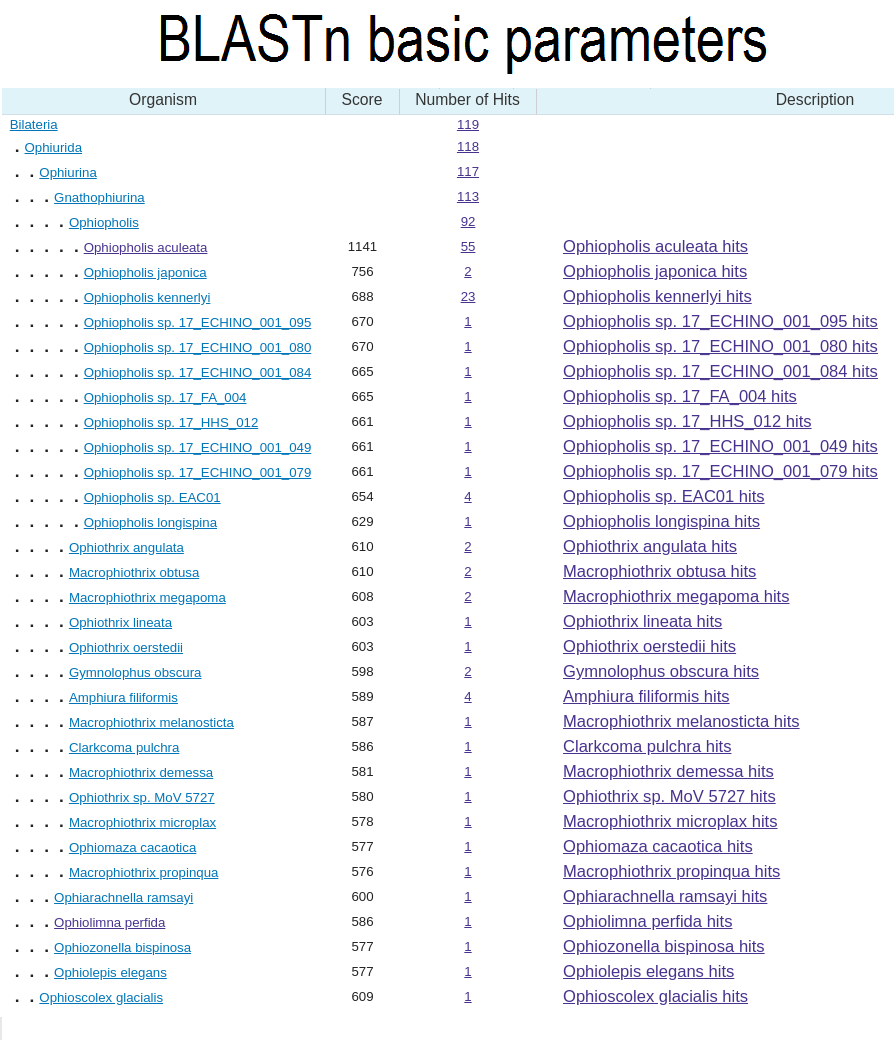

А вот пример с вирусом оказался более интересным: известно, что у вирусов вариативность генов больше. Входными данными работы алгоритмов была одна из последовательностей CDS.
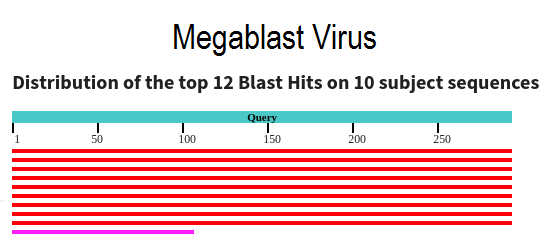
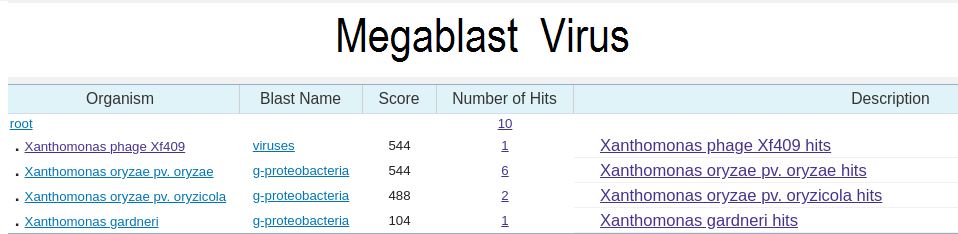

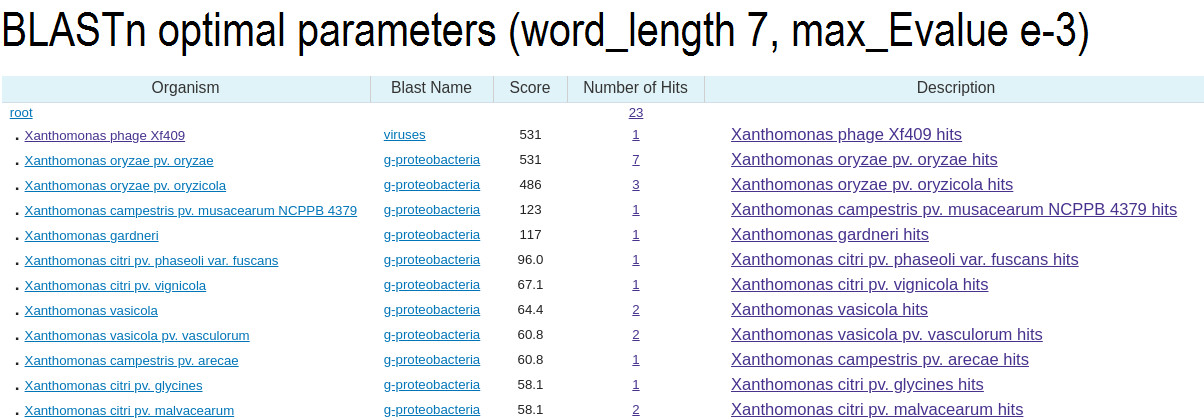
Megablast выдал мало последовательностей (10). Он настроен на поиск очень близких последовательностей, а так как геномы вирусов салбо консервативны, то найти очень близких гомологов последовательности трудно.
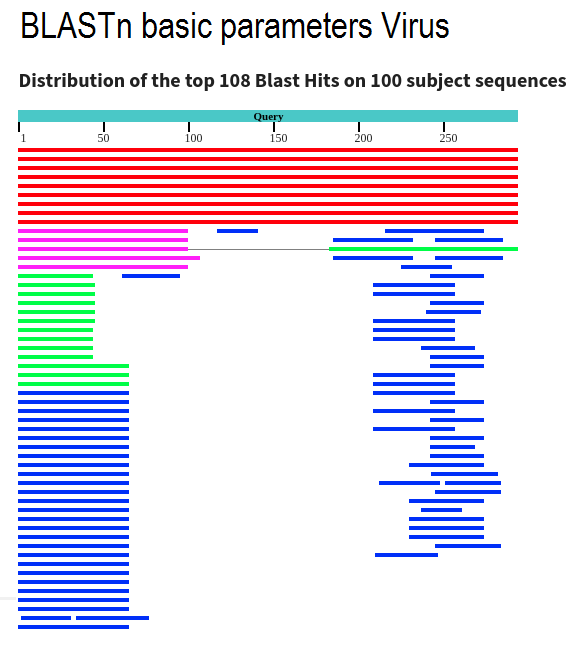
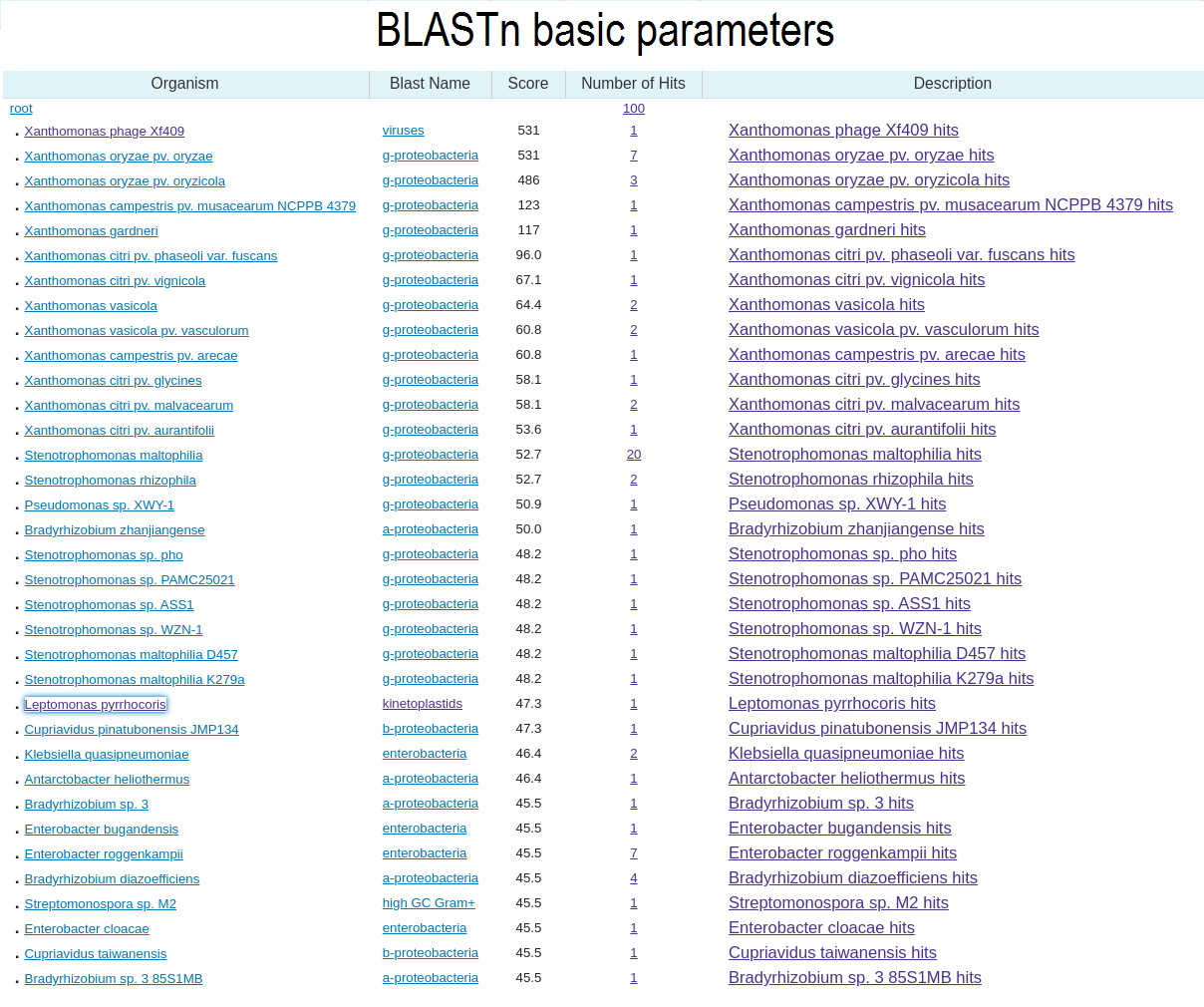
Blastn с параметрами по умолчанию нашел много последовательностей (вывел 100), которые хотя бы частично похожи на введенную. При этом весы выравнивания абсолютного большинства выведенных последовательностей меньше 100, что очень плохо. На основании этого оптимальные параметры выбирались, чтобы отмести выравнивания с плохим Score (Evalue < 0.001, длина слова 7, на случай если blastn что-то пропустил, число выведенных последовательностей 50 - не повлияло, их и так меньше вывелось). При этих параметрах вывелось 23 последовательности, выравнивания половины из них очень хорошие, еще четверти - просто хорошие (по длине и качеству выравнивания). Можно сказать, что из этих 3 алгоритмов лучше всего с вирусами сработал blastn с подобранными параметрами.
Task 3
По заданию требовалось научиться принципам работы с пакетом программ blast+, позволяющим работать с BLAST через терминал. Было предложено задание определить наличие или отсутствие трех белков, которые предположительно есть у всех эукариот, в геноме организма Amoeboaphelidium protococcarum, принадлежащего группе близкой грибам. В связи с этим нами на последовательности белков для проверки blast+ накладывались следующие требования:
- Белок связан с процессами или структурами, специфичными для эукариот;
- Последовательность берется из протеома какого-нибудь гриба;
- Последовательность должна быть высоко консервативной.
Нами были выбраны 3 белка аскомицет:
- H2B, 148 aa (Talaromyces cellulolyticus - одна из множества телеоморф пеницилла) - один из гистонов эукариот. Как известно, нуклеосомная организация генетического материала характерна для эукариот и некоторых архей, но у архей гистоны совершенно другие.
- U5, 988 aa (Uncinocarpus reesii - сапротрофный грибок, живущий на кератиновых материалах) - компонент малой субъединицы сплайсосомы, обеспечивающий распознавание 3'-сайта сплайсинга на одном из этапов. Сплайсинг - процесс, характерный для эукариот.
- SUI1, 108 aa (Saccharomyces cerevisiae - без комментариев) - фактор инициации трансляции eIF1, повышающий сродство более функциональных факторов инициации трансляции и субъединиц рибосом. У эукариот инициация трансляции отличается от таковой у пркариот, и ф.и.т. называются по-другому (eIF против IF).
Для проверки наличия белка использовалась программа tblastn, транслирующая базу данных и выравнивающая ее с последовательностью. База данных - геном Amoeboaphelidium protococcarum, лежащий в файле 'Genome.fasta'. В качестве последовательностей брались файлы с последовательностями соответствующих белков, полученных с разных сайтов.
Строка, с помощью которой выполнялась программа:
tblastn -query name.fasta -db Genome.fasta -out name.out -evalue 0.01 -outfmt 7
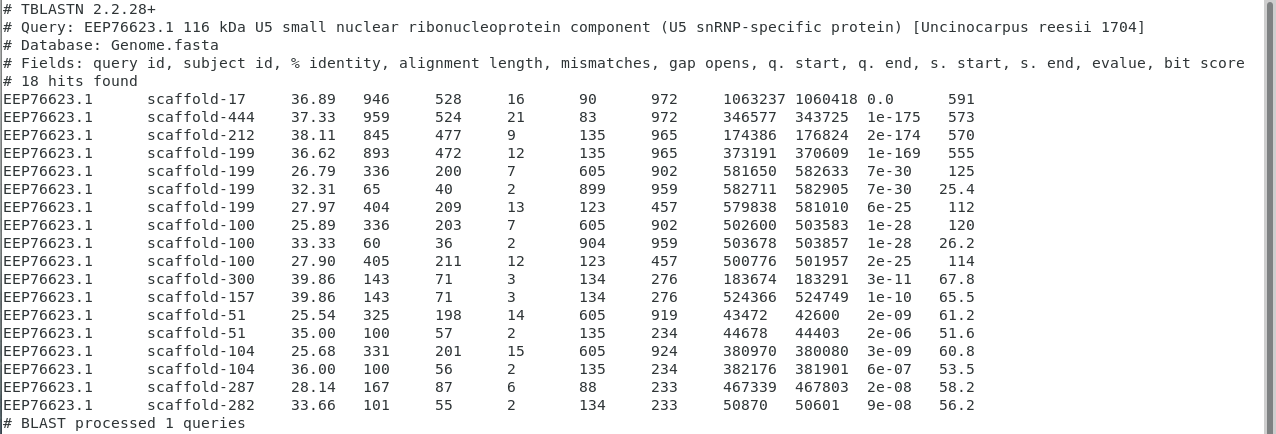
Таким образом были получены три файла: H2B.out, U5.out, SUI1.out. Содержимое файлов - на картинках.


Как можно видеть, H2B гриба хорошо выравнялся с одним из скаффолдов (evalue e-41, Identity 78%), но покрытие 96/148 = 64.86%, не очень хорошее: первые 50 аминокислот вообще не покрыты. Для проверки в blastp на сайте NCBI была проверена последовательность, и оказалось, что все показанные выравнивания (evalue не больше 1e-40) связаны с последовательностями H2B других организмов.
У белка U5 все сложнее: evalue очень хорошие, покрытие замечательное (примерно 953/988 = 96.46%), но Identity 35-40%. При проверке blastp оказалось, что не все последовательности с такими параметрами выравнивания относятся к U5 (были факторы элонгации, например), а значит утверждать наличие белка на основании этих данных мы не можем. Для дальнейшей проверки нужно выровнять последовательность из генома исследуемого организма с глобальной базой данных. Есть вероятность, что в геноме просто содержатся белки с такими же доменами, что и наш фактор элонгации.
С белком SUI1 ситуация гораздо лучше (Identity 56%, evalue малы, покрытие 105/108 = 97.22%). При проверке blastp выяснилось, что все последовательности с примерно такими параметрами и лучше принадлежат этому фактору инициации трансляции, а знаячит можно с очень высокой вероятностью считать, что этот белок у исследуемого организма есть.
Task 4
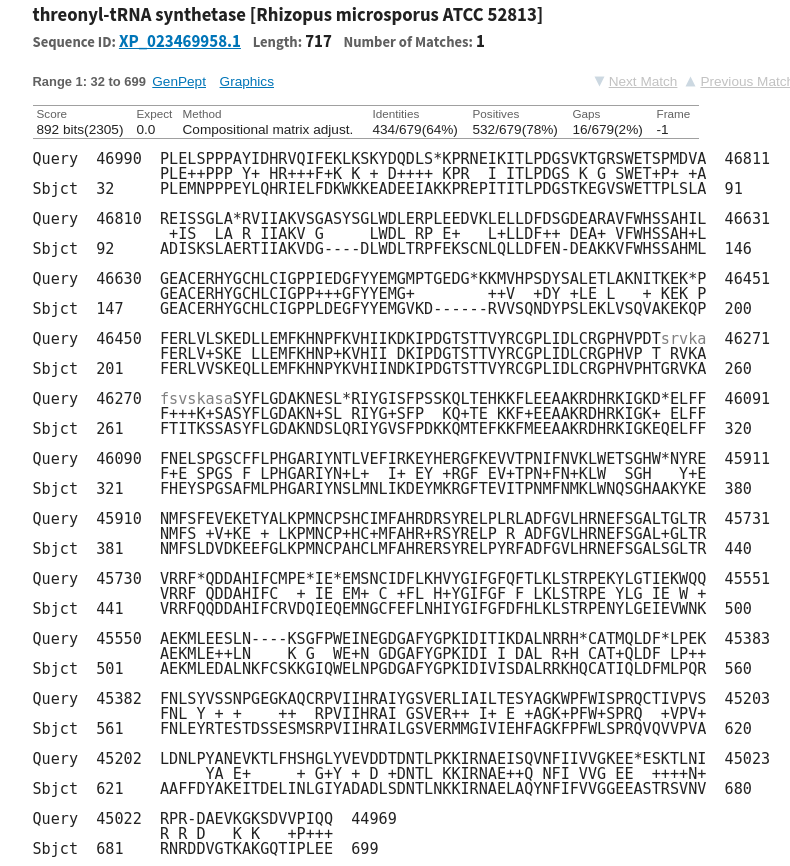
По заданию мы нашли какой-то скаффолд (а именно 687) в геноме исследуемого в предыдущем задании организма (родственного грибам) с длиной порядка десятков тысяч нуклеотидов (61498 в данном случае). Требовалось найти какой-то ген в этом скаффолде. Мы использовали для этого blastx, выравнивающий транслированный скаффолд с базой данных белков (Refseq Protein, мы так ограничили поиск). Кроме базы данных, ограничения коснулись таксонов происка (ограничили таксоном Fungi). BLASTx выдал какие-то последовательности, большинство из последовательностей в верхних строчках относились к белку треонил-тРНК синтетазы. Evalue их равны 0, Identity 60-65%, вес более 850. На основании этого можно сказать, что наш скаффолд содержит ген этого белка. Вот параметры одного выравнивания для примера: