Ресеквенирование. Поиск полиморфизмов у человека
Подготовка чтений
Были даны два файла 22 хромосомы человека: -fastq (файл с последовательностями и качеством для каждого нуклеотида; разделителем строки качества и строки последовательности является '+') и -fasta формата (обычный fasta-файл). Для первого задания будем использовать только fastq-файл.
Сперва необходимо проверить качество 'сырого' файла. Для этого воспользуемся программой FastQC, а именно следующей командой:
fastqc chr22.fastq
В итоге получаем архив и html-файл (картинки с объяснением будут ниже). Чтобы работать с наиболее корректными данными воспользуемся программой Trimmomatic, которая сможет исключить некоторые огрехи, такие как: короткая последовательность, низкокачественные чтения... Команда, которая будет удалять чтения менее 50 нуклеотидов и чтения качества меньше 20, следующая:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr22.fastq chr22_trim.fastq TRAILING:20 MINLEN:50
Также, неплохо было бы показать, что действия с Trimmomatic имеют смысл, поэтому очищенный файл прогоняем через FastQC и получаем на выходе архив и html-файл, которые будем сравнивать с исходными.
Итак, сравним данные, полученные программой FastQC до использования Trimmomatic и после:
| Таблица 1. Сравнение некоторых параметров выдачи программой FastQC | ||
|---|---|---|
| 'Сырые' данные | Данные после обработки Trimmomatic | |
| Количество последовательностей | 11.427 | 11.091 |
| GC, % | 51 | 50 |
*Стоит отметить, что содержание N (условное обозначение любого нуклеотида) изначально равнялось 0, как и содержание адаптеров.
Приведенные выше результаты не говорят о реальной разнице, которую могут показать такие параметры, как Per base sequence quality и Per sequence quality scores. Ниже представлено сравнение этих двух параметров:
 |
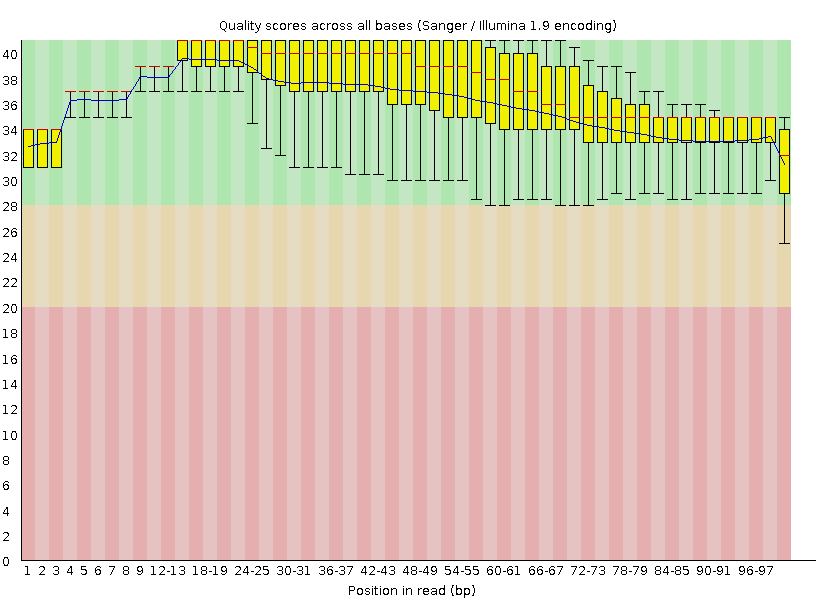 |
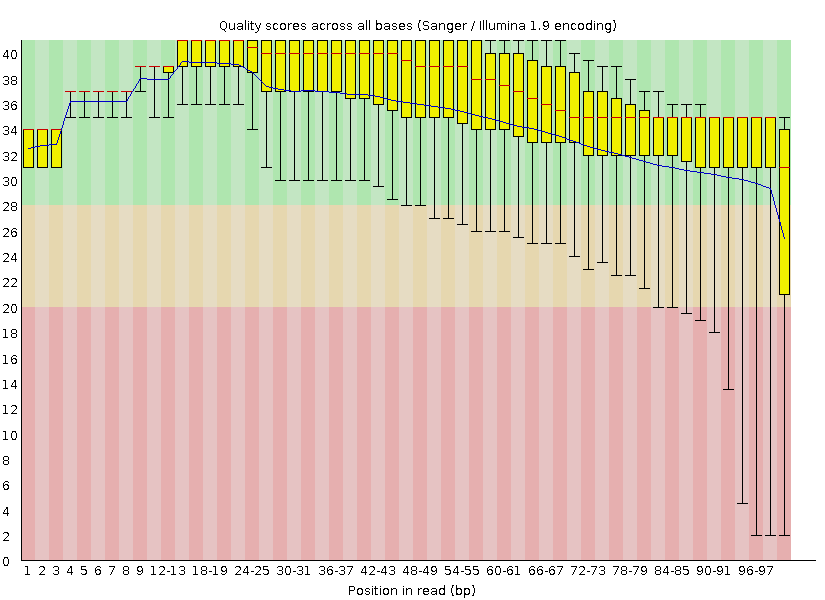
На картинке сверху слева представлен график Per base sequence quality для исходного файла, а справа - для обработанного. По оси Y отложен score, а по оси X - позиция в риде (bp). Видно, что качество повысилось довольно сильно на конце (из-за использования комбинации функций MINLEN и TRAILING), чего нельзя сказать про середину рида, хотя качество уже в исходном файле не опускалось ниже 34, что соответствует более 99.9% точности.
 |
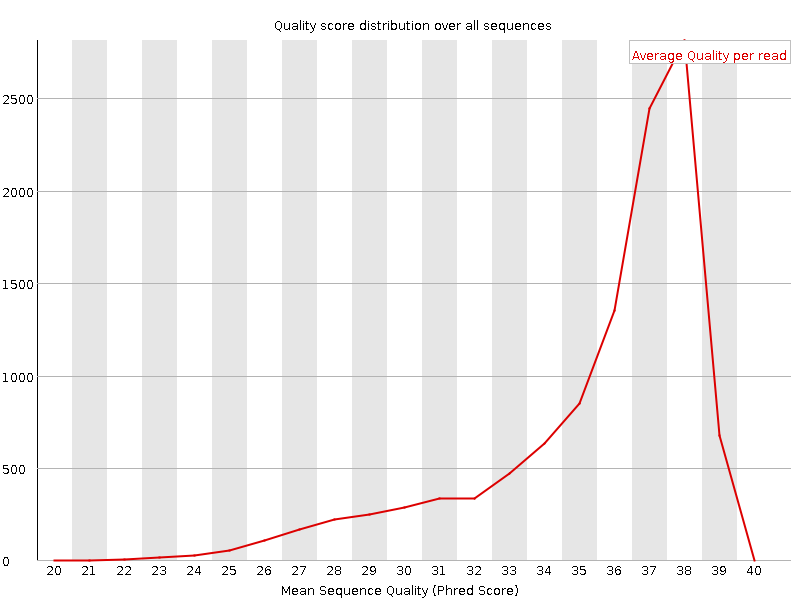 |
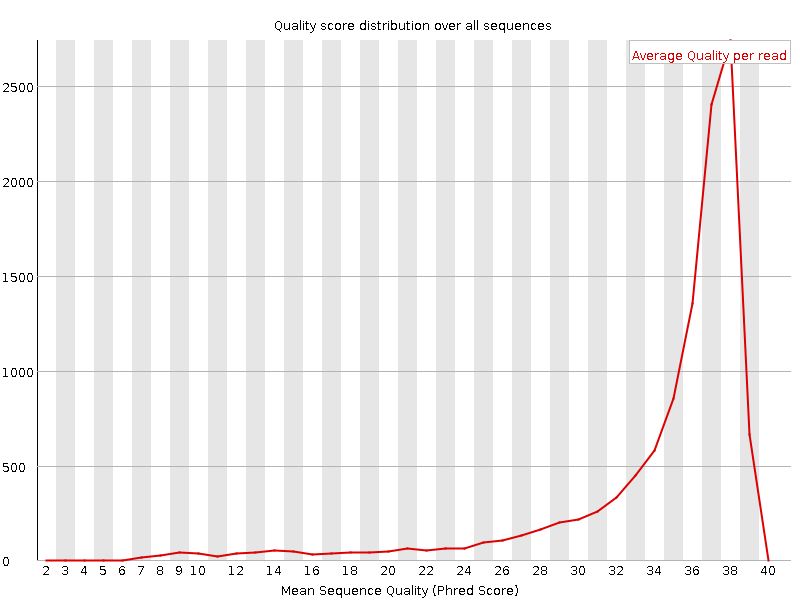
Мне показалось, что графики, представленные выше (сверху слева для исходного файла и справа для обработанного), характеризующие Per sequence quality scores, также неплохо иллюстрируют изменения в качестве рида.
По оси Y на графиках отложено количество последовательностей, по оси X - среднее значение качества последовательности. Хорошо видно, что на графике слева (исх.) значения на оси X начинаются с 2, тогда как на графике справа (обр.) - с 20 (количество ридов уменьшилось, причем за счет уменьшения низкокачественных). Пик соответствует среднему значению качества на рид, который как до, так и после обработки остался на значении 38 единиц (более чем хорошее качество). К слову, если среднее опускается ниже 28, то программа выдает Warning, а если 20 - Failure.
Картирование чтений
При помощи программы BWA были откартированы очищенные чтения. Это было сделано в два этапа:
1) Индексирование референсной последовательности:
bwa index chr22.fasta
2) Построение выравнивания референса и прочтений:
bwa mem chr22.fasta chr22_trim.fastq > chr22.sam
Далее необходимо было проанализировать выравнивание, посчитать количество откартировавшихся чтений на референсную последовательность. Вот шаги этого многостадийного процесса:
1) Перевод выравнивания в бинарный формат:
samtools view chr22.sam -b -o chr22.bam, где флажок -b означает то, что расширение выходного файла изменено
2) Сортировка выровненного файла с референсом по координате в референсе начала чтения:
samtools sort chr2.bam -T trash.txt -o chr22_sort.bam, где флажок -T означает запись в некоторый временный файл, а не в stdout
3) Индексация отсортированного .bam-файла:
samtools index chr22_sort.bam
4) Выяснение количества откартировавшихся чтений:
samtools idxstats chr22_sort.bam > cart.out
В итоге откартировались 11090 чтений, 2 не откартировались.
Далее был найден один экзон со средним покрытием. Это было сделано следующим образом:
Был сделан файл с числом покрытий каждого нуклеотида:
samtools depth chr22_sort.bam > depth.out
Далее при помощи excel (фильтр по столбцу с количеством ридов и некоторых простых команд) я заметил, что хорошим покрытием можно считать нуклеотид с числом ридов более 100. Был выбран нуклеотид 28.379.346 (число ридов - 110). На сайте NCBI я выбрал эту версию сборки генома человека, где в геномном браузере смотрел 22 хромомосому. В итоге на картинке можно заметить, что нуклеотид попал точно в экзон, для которого характерен альтернативный сплайсинг.
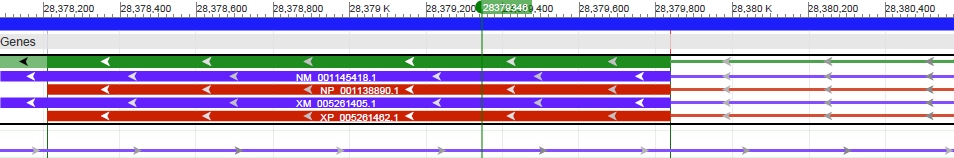
Координаты этого экзона - 28.378.208-28.379.839(r). При помощи команды samtools был получен этот файл:
samtools depth chr22_sort.bam -r chr22:28378208-28379839 > depth_exon.out
Чтобы посчитать среднее был написан скрипт, который в итоге вывел число 52, которое соответствует среднему покрытию.
Анализ SNP
Первым шагом стало создание файла с полиморфизмами в формате .bcf:
samtools mpileup -uf chr22.fasta chr22_sort.bam -o snp.bcf
Вторым шагом стало создание файла со списком отличий между референсом и чтениями в формате .vcf:
bcftools call -cv snp.bcf -o snp.vcf
Чтобы показать интересные отличия, была составлена таблица ниже:
| Таблица 1. Примеры полиморфизмов | |||||
|---|---|---|---|---|---|
| Тип полиморфизма | Позиция | В референсной последовательности | В последовательности с чтениями | Качество | Глубина покрытия |
| Вставка | 26.159.129 | CTTT | CTTTTT | 154.468 | 9 |
| Замена | 26.164.079 | C | T | 221.999 | 39 |
| Делеция | 26.163.823 | gt | g | 30.2276 | 2 |
Всего было найдено 14 INDEL и 214 полиморфизмов.
*Все три SNP, которые представлены в таблице, входят в экзоны белка миозина 18B (MYO18B).
**Интересно, что абсолютно все (214) SNP располагаются в 4 генах: миозин 18B (MYO18B), тетратрикопептид повторяющийся домен (TTC28), антисмысловая РНК (TTC28-AS1), которая кодируется некодирующими РНК (ncRNA), и аполипопротеин L1 (APOL1).
Также хотелось бы добавить, что этот тетратрикопептид повторяющийся домен принимает участие в клеточном делении (ссылка); аполипопротеин характерен только для некоторых видов обезьян и человека, входя в сложный комплекс липопротеина высокой плотности (ЛПВП) (ссылка).
***Качество представленных полиморфизмов в целом хорошее, хотя делеция обладает довольно низким качеством относительно замены и вставки.
Далее все INDEL были удалены и файл только с заменами создан. Для того, чтобы программа annovar смогла работать с файлом, была использована следующая команда:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_out.vcf -outfile snp.annovar
Уже непосредственно с полученным файлом и будем работать дальше.
1) Аннотация по базе refgene:
perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr22.refgene -build hg19 snp.annovar /nfs/srv/databases/annovar/humandb/
На выходе три файла: файл с описанием работы (.log), файл с описанием всех SNP (.variant_function), файл с описанием SNP, попавших в экзоны (.exonic_variant_function).
В итоге 181 SNP находятся в интронах, 26 - в экзонах и 7 в некодирующей части РНК (UTR), при этом 75 замен гетерозиготных и 139 гомозиготных. Эти результаты ожидаемы, так как бОльшее количество замен логичнее ожидать в интронах (частях, не кодирующих белки), так как они не кодируют белки, поэтому замена последовательности нуклеотидов не повлечёт за собой замену последовательности белка, изменение их функции и т.д.
2) Аннотация по базе dbsnp:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr22.dbsnp -build hg19 -dbtype snp138 snp.annovar /nfs/srv/databases/annovar/humandb/
На выходе три файла: файл с описанием работы (.log), файл (1), в котором указаны SNP, аннотированные в dbSNP (rs) и файл (2), в котором указаны SNP, не аннотированные в dbSNP. В (1) файле содержатся 176 SNP (входят в кластер rs), в (2) - 38, причем качество последних очень плохое (не превышает 33).
3) Аннотация по базе 1000 genomes:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out chr22.geno snp.annovar /nfs/srv/databases/annovar/humandb/
На выходе три файла: файл с описанием работы (.log), файл (1'), в котором указаны SNP, аннотированные в 1000genomes (их частота встречаемости) и файл (2'), в котором указаны SNP, не аннотированные в 1000genomes. В (1') файле содержатся 171 SNP, в (2') - 43, причем в отличие от прошлой базы данных нельзя сказать, что качество SNP в файле (2') очень плохое.
Также на рисунке ниже представлена гистограмма распределения SNP по частотам встречаемости:
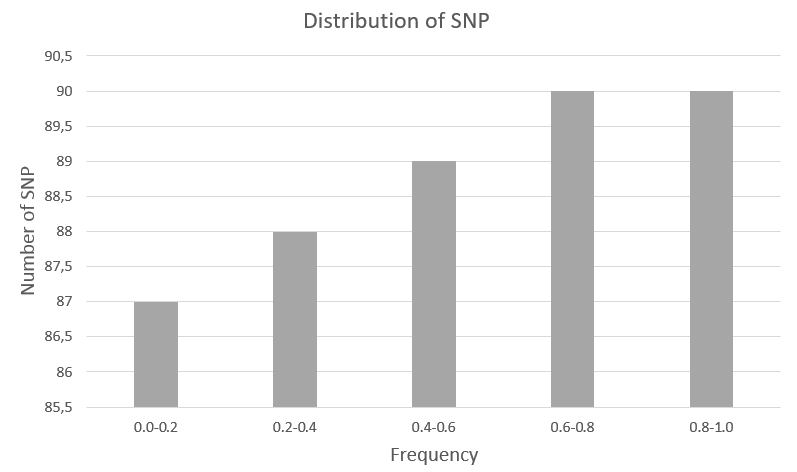
Видно, что в целом частота встречаемости аннотированных в 1000genomes SNP распределена равномерно от значений 0.0-0.2 до 0.8-1.0. Небольшим увеличением (на 3) количества полиморфизмов можно пренебречь.
4) Аннотация по базе GWAS:
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr22.gwas -build hg19 -dbtype gwasCatalog snp.annovar /nfs/srv/databases/annovar/humandb/
На выходе 2 файла: файл с описанием работы (.log), файл с описанными клиническими случаями для SNP. Интересно, что в файле лежали 3 SNP, причем одна из них связана с математическими способностями у детей с дислексией (!), другая - с ожирением, а третья - с гломерулосклерозом.
5) Аннотация по базе :
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr22.clincar -buildver hg19 -dbtype clinvar_20150629 snp.annovar /nfs/srv/databases/annovar/humandb/
На выходе 3 файла: файл с описанием работы (.log), файл(1'') с аннотированными SNP, файл(2'') со всеми остальными SNP. В (1'') файле лежат 2 SNP, оба связаны с гломерулосклерозом, во (2'') файле - все остальные SNP.
{ссылка на итоговую таблицу со всеми SNP и их краткой характеристикой}
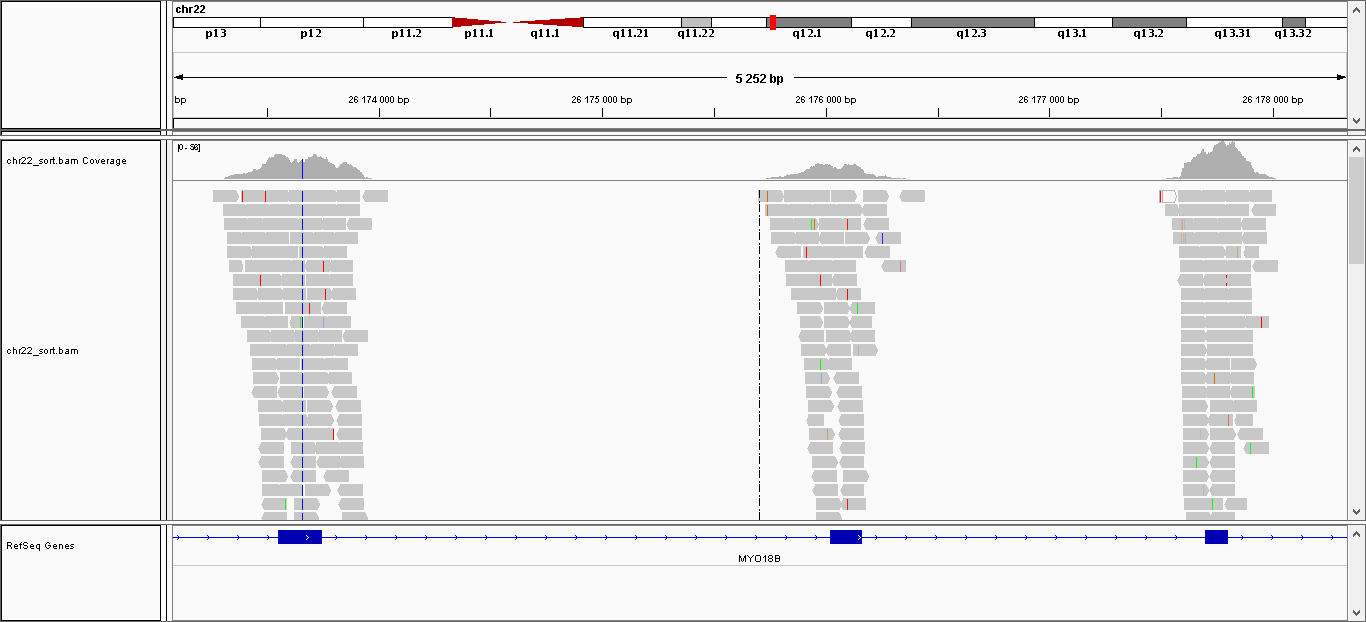
Последним заданием была визуализация трех SNP в IGV. Для этого на вход были даны два файла: отсортированный .bam-файл и индексированный файл для .bam-файл. В итоге были получены следующие картинки:
Скрин для вставки (видны инсерции в 4 ридах);
{kind=link}
Скрин для замены (только в 1 риде нет замены);
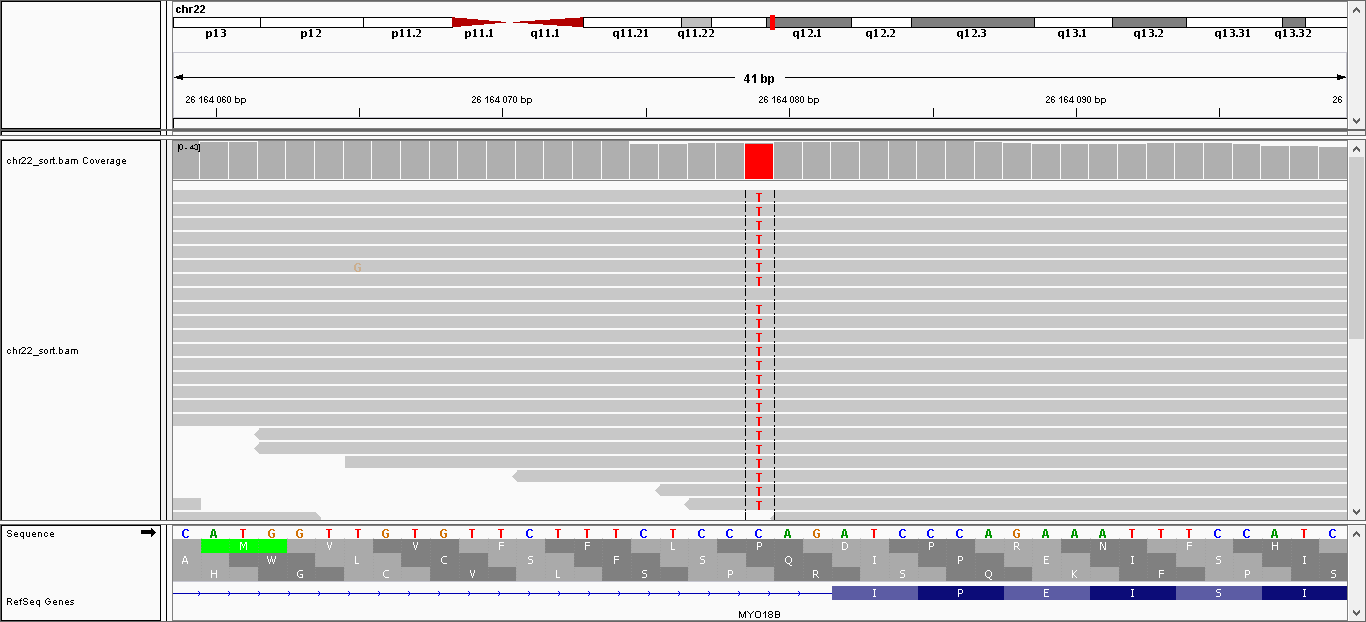{kind=link}
Скрин для делеции (в обоих ридах есть делеции).
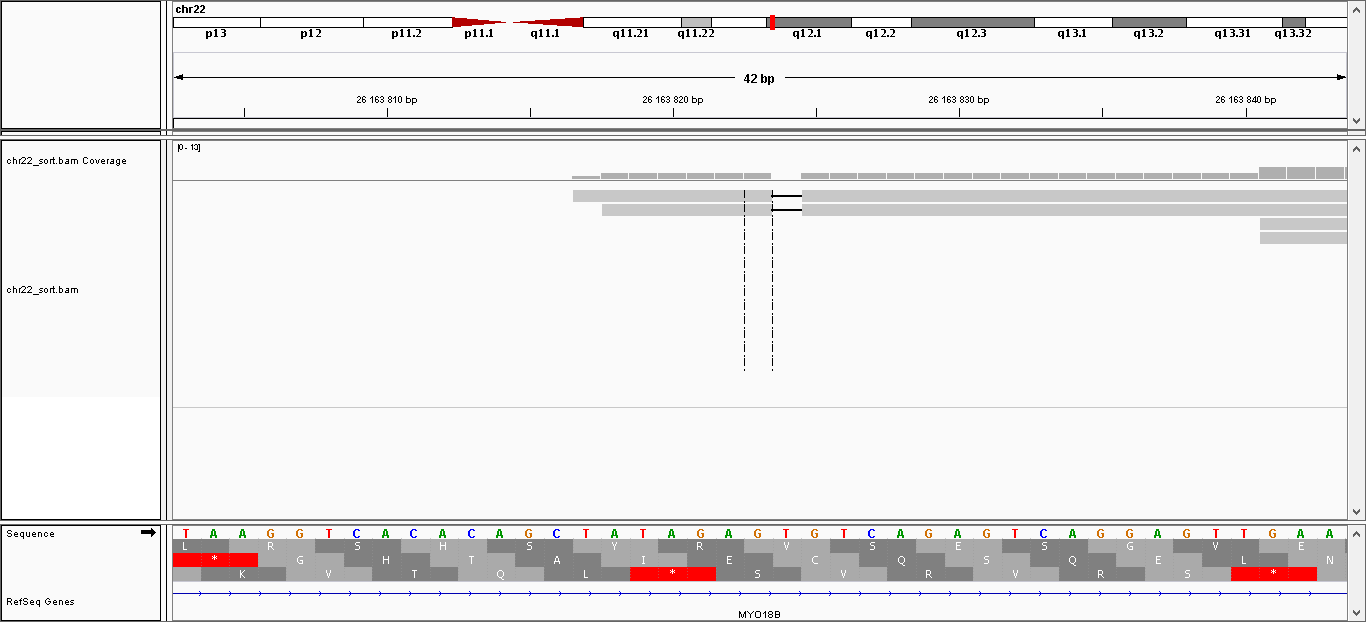{kind=link}
К сожалению скрины получились слишком большими, поэтому только ссылки :(. Что касается относительного расположения ридов и экзонов в референсной последовательности, то можно утверждать, что риды довольно хорошо ложатся на экзоны (например).
{kind=link}
⌘
© Emir Radkevich, 2016