Чтение последовательностей по Сэнгеру
Чтение заданной последовательности
{Ссылка на исходный файл с прямой цепью}
{Ссылка на исходный файл с обратной цепью}
Изначально были даны два файла с расширением .ab1, которые нужно было проанализировать с помощью программы Chromas. Чтобы корректно проанализировать выдачу секвенатора, следовало воспользоваться функцией Reverse+Complement для обратной цепи и сравнить две последовательности. Длина нечитаемых концов разнится у цепочек, что позволило предугадать некоторые неидентифицированные нуклеотиды (N) в одной из последовательностей с помощью другой.
Следующая таблица показывает координаты нечитаемых концов каждой цепочки:
| Таблица 1. Координаты нечитаемых концов каждой цепочки | ||
|---|---|---|
| Направление конца последовательности | Прямая цепь | Обратная цепь |
| 5'- | 1-17 | (1-8)/- |
| 3'- | 691-702 | 683-716 |
Несмотря на то, что некоторое число нуклеотидов оказалось нечитаемо, в целом, качество хроматограммы хорошее. Уровень шума относительно интенсивности сигнала нуклеотида небольшой и не сильно колеблется по всей длине хроматограммы.
Что касается уровня самого сигнала, то на картинках ниже можно увидеть небольшое различие в интенсивности сигнала.
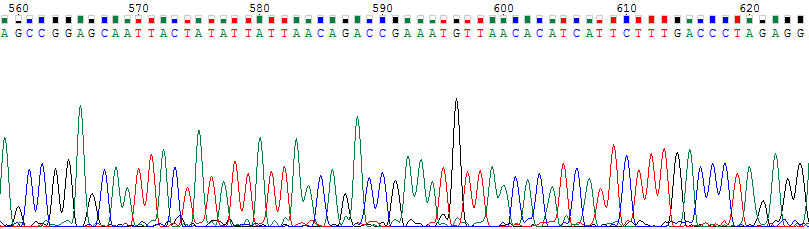
Рис.1. Слабый сигнал нуклеотидов 621G (27 единиц) и 560G (17 единиц).
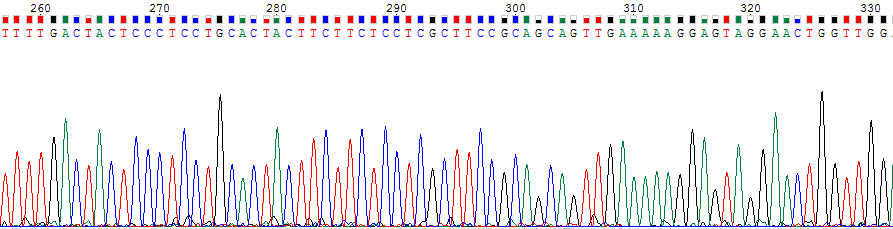
Рис.2. Сильный сигнал нуклеотидов 326G (51 единица) и 276G (46 единиц).
Прежде чем сохранить последовательности в fasta-формате, необходимо было устранить максимально возможное устранимое количество сложных мест в обеих последовательностях.
Ниже будут перечисляться некоторые разрешившиеся пробленые места с пояснениями на картинках.
Сложности
Сложность 1 - возможный полиморфизм.
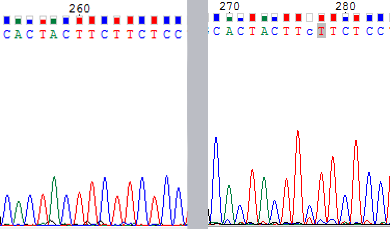
Рис.3. На картинке справа (от серой линии) в позиции 278 наблюдается явная возможность полиморфизма, которая однако исчезает, если взглнуть на комплементарную цепь, где четко видно, что в позиции 262 (картинка слева) стоит C.
Сложность 2 - сдвиг пиков.
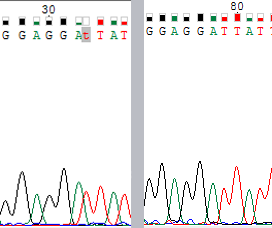
Рис.4. В позиции 33 на картинке слева (от серой линии) четко виден сдвиг пика, несмотря на то, что сигнал достаточно сильный. При сравнении с комплементарной последовательностью было выяснено, что в позиции 33 (картинка слева) находится T.
Сложность 3 - расширение пика и резкое увеличение интенсивности сигнала.
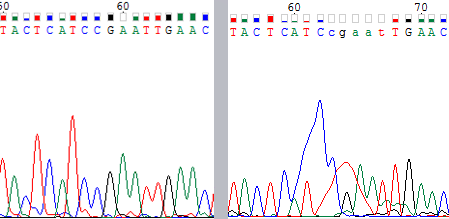
Рис.5. Одна из наиболее сложных ситуаций представлена на этой картинке. Целый блок нуклеотидов нечитабелен (картинка справа от серой линии). Однако, после сопоставления с комплементарной цепочкой (картинка слева) можно с некотрой увереностью написать нечитаемую последовательность. Утверждать такое, можно только потому, что окружение этой последовательности и сама последовательность на комплементарной цепочке отсеквенирована с почти идеальной точностью (расстояние между пиками в широком диапазоне одинаковое, отношение интенсивности сигнала к шуму высокое).
В итоге были получены следующие последовательности:
Ссылка на fasta-файлы с прямой и обратной цепями.
Выравнивание двух цепей, полученных с помощью секвенатора
С помощью Jalview был получен проект, в котром показано выравнивание двух последовательностей.
На картинке снизу также можно видеть результаты:

Рис.6. Выравнивание двух цепочек, полученных с помощью секвенатора
Как видно внизу картинки, была создана группа 'Error nucleotide', показывающая буквой 'E' нуклеотиды, которые были исправлены в ходе сопоставления с комплементарной последовательностью, и буквой 'P' возможные полиморфизмы, которые не на самом деле не являлись таковыми.
Пример нечитаемого фрагмента
Для того, чтобы продемонстрировать нечитаемый фрагмент, была взята концевая часть данной мне последовательности.
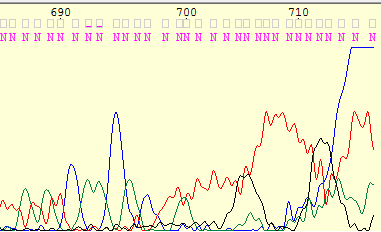
На рисунке выше видно, что идентифицировать какой-либо нуклеотид практически невозможно. На этом фрагменте хорошо различимы сразу несколько проблем:
* расстояние между пиками на всей протяженности фрагмента неодинаково;
* не всегда удается различить шум от пика;
* 'пятно' (резкое возрастание интенсивности сигнала);
* наложение одного пика на другой (псевдополиморфизм).
Исходя из вышесказанного, несложно понять, что такой фрагмент нельзя интерпретировать ни в коем случае!
⌘
© Emir Radkevich, 2016