Нуклеотидный BLAST
Таксономия и функция прочтенной последовательности
В практикуме 6 был получен fasta-файл с последовательностью нуклеотидов. При стандартных параметрах был запущен алгоритм blastn по базе данных nucleotide collection. На выходе было получено следующее (показаны первые 10 находок):
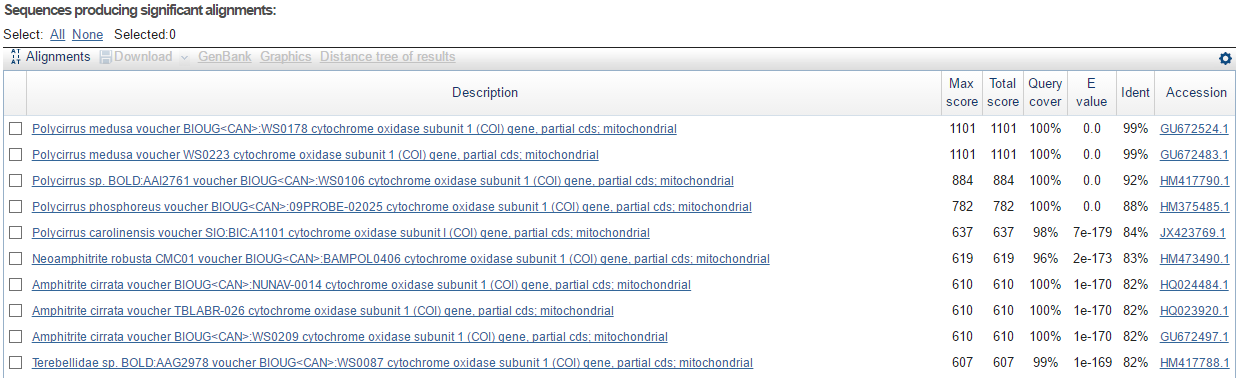
Несложно понять, что прочтенная последоватенльность является митохондриальным геном многощетинкового червя Polycirrus medusa. Этот ген кодирует субъединицу 1 цитохром с-оксидазы. Этот фермент катализирует реакцию переноса с цитохрома с на кислород с образованием воды в аэробной дыхательной цепи переноса электронов. Субъединица 1 является главным компонентом цитохром с-оксидазы, несет на себе основные кофакторы и осуществляет основные реакции катализа.
Чтобы удостовериться в правильности выбора организма, было скачано несколько последовательностей, находящихся в топе выдачи blast, и выровнено с помощью алгоритма muscle в jalview. Результаты выдачи программы можно видеть ниже:

На вход подавалась последовательность '1947'. Видно, что все последовательности достаточно похожи, однако наибольшее сходство наблюдается с последовательностью гена, кодирующего цитохром с-оксидазу, Polycirrus medusa.
{Ссылка на jvp-проект}, {Ссылка на fasta-файл с последовательностями}
P.S.: Систематика Polycirrus medusa начиная с царства до рода: Metazoa; Eumetazoa; Bilateria; Protostomia; Lophotrochozoa; Annelida; Polychaeta; Scolecida; Terebellida; Terebellidae; Polycirrus
Сравнение списков находок нуклеотидной последовательности разными алгоритмами blast
В сравнение участвовали три алгоритма: discontiguous megablast, megablast и blastn. Чтобы выдача была более-менее показательна были изменены следующие опции:
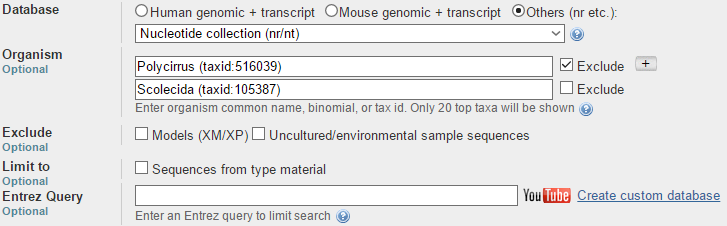
Как видно, исключались виды рода Polycirrus, и при этом поиск шел внутри подкласса Scolecida. Что касается количества находок, то для разных алгоритмов он был разным. В таблице ниже отражены все основные характеристики по двум алгоритмам:
| Таблица 1. Сравнение алгоритмов | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Алгоритм | Количество находок | Лучший score | Худший score | Лучший E-value | Худший E-value | Лучший ident, % | Худший ident, % | Лучшее покрытие, % | Худшее покрытие, % |
| Megablast | 14 | 547 | 156 | 4e-156 | 3e-38 | 83 | 74 | 100 | 64 |
| Blastn | 1000 | 619 | 486 | 7e-178 | 1e-137 | 83 | 78 | 100 | 92 |
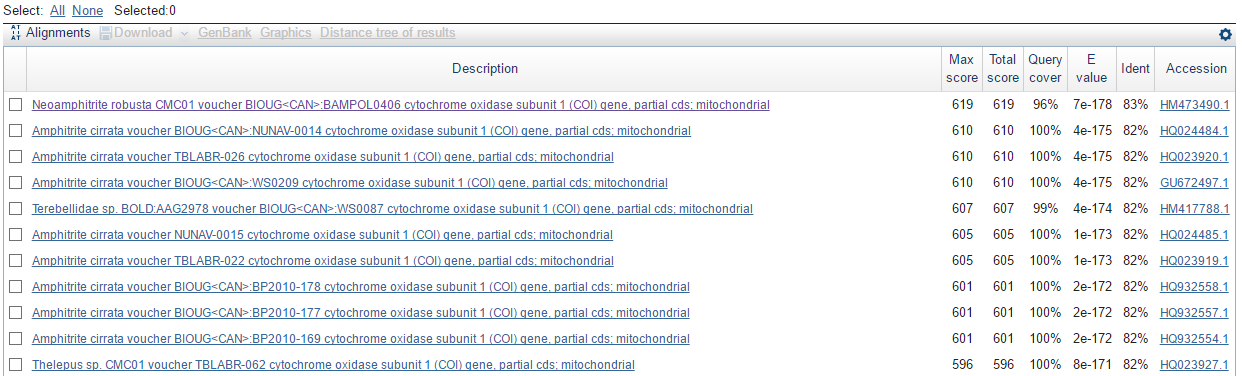
В таблице приведены только два алгоритма. Это связано с тем, что выдача discontiguous megablast была идентична выдаче blastn, по крайней мере до тех пор, пока размер слова (word size) не был изменен. Что касается самой выдачи, megablast, как это видно, более тщательно отбирает находки, то есть короткие последовательности могут и не быть в выдаче, только наиболее похожие последовательности показываются в итоге.
Ниже можно посмотреть на 10 лучших и худших находок blastn:

А также на все находки megablast:
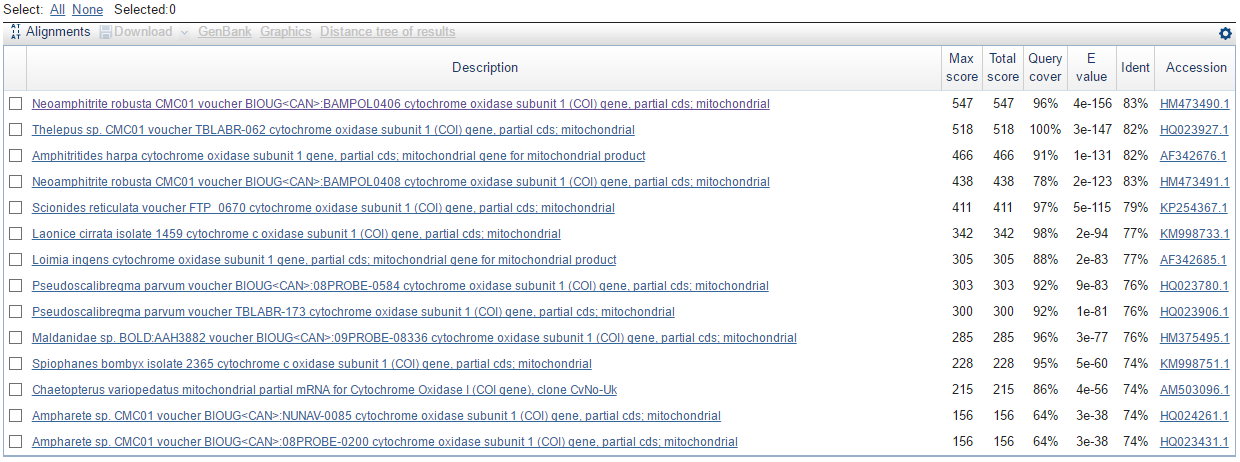
Стоит отметить, что megablast нашел некоторые последовательности, которые не нашли discontiguous megablast и blastn: Loimia ingens, Pseudoscalibregma parvum, Spiophanes bombyx и некоторые другие. На самом деле это неудивительный результат, так как изначально blastn был ограничен 1000 последовательностями. Если расширять этот диапазон, то будут появляться организмы, о которых сказано двумя строчками выше (такое было проделано при ограничении в 20000 последовательностей).
Подводя итоги, можно сказать, что при данных параметрах blastn и discontiguous megablast работают схоже, на их выходе получается бОльшее колчичество последовательностей, чем на выходе megablast, что объясняется более строгим отбором в алгоритме megablast. При изменении параметра word size в алгоритме discontiguous megablast сам алгоритм выдает наибольшее количество последовательностей среди всех алгоритмов.
Наличие гомологов белков в организме X5 (Amoebaphelidium protococcarum)
Необходимо было проверить наличие гомологов определеных белков в организме X5. Для этого была создана локальная база данных из X5.fasta и с помощью tblastn находились возможные гомологи (скэффолды в fasta-файле X5 в данном случае выступали в роли возможных гомологов).
В дальнейшем будут рассматриваться белки со следующими prot ID: TERT_HUMAN, RPB1_HUMAN, PABP2_HUMAN.
На картинке ниже представлены теоретически гомологичные скэффолды белку TERT_HUMAN, или теломераза, восстанавливающая длину хромосомы:

Видно, что E-value достаточно высокий, а score небольшой у scaffold-17, однако нельзя отрицать, что белок, транскрибирующийся со скэффолда-17 вполне может оказаться гомологичным белку TERT_HUMAN.
Следующая картинка отражает тоже самое только для белка RPB1_HUMAN, или субъединица ДНК-зависимой РНК-полимеразы II:

Очевидно, что scaffold-300 и scaffold-157 являются гомологичными белку RPB1_HUMAN. На это указываеют очень большое значение score, а также E-value, равный 0.0.
Выдача tblastn для белка PABP2_HUMAN:
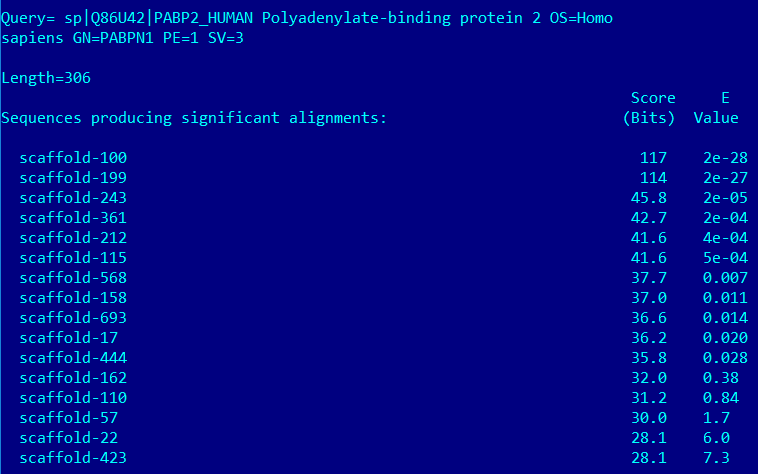
Аналогичная ситуация уже наблюдалась с белком TERT_HUMAN: scaffold-100 и scaffold-199 могут оказаться гомологичными белку PABP2_HUMAN. Интересно отметить, что scaffold-100 уже был замечен, как теоретически возможный гомолог белку RPB1_HUMAN, причем с бОльшим значением score и меньшим занчением E-value.
Поиск гена, закодированного в одном скэффолде Amoebaphelidium protococcarum
Был выбран скэффолд подходящей длины - scaffold-700. Fasta-файл с последовательностью скэффолда выступал в качетсве входного файла для blastx. Ниже представлена картинка с лучшими результатами выдачи программы:
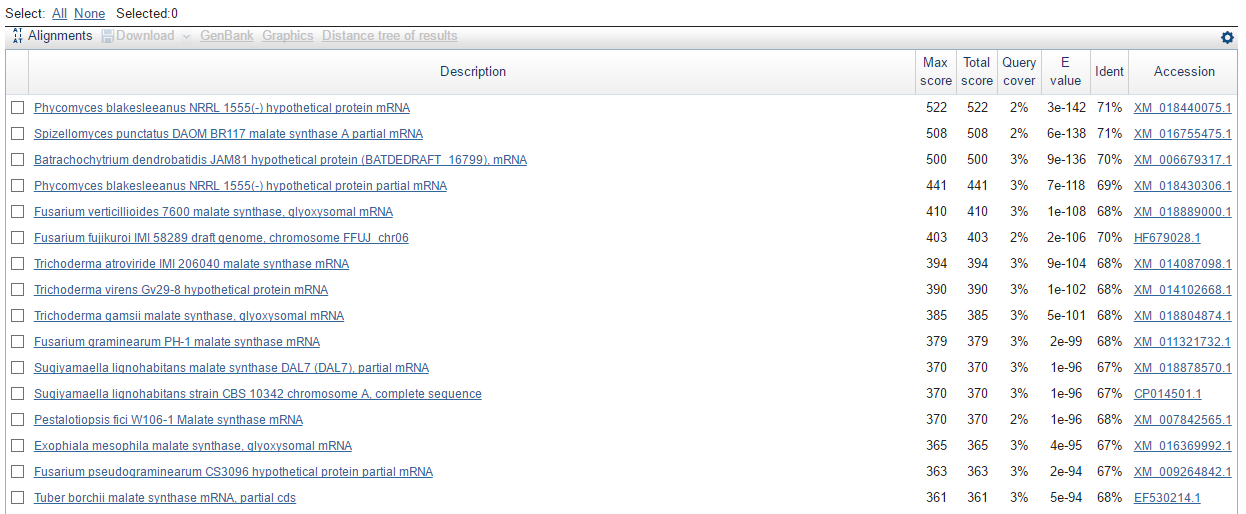
Видно, что небольшая часть (около 2%) скэффолда вероятнее всего кодирует фермент малат-синтазу, фермент катализирующий трансферазную реакцию (КoA отщепляется от ацетил-КоА, при этом образованный продукт соединяется с глиоксилатом и водой с образованием малата).
Классифкация геномов родственных вирусов по сходству последовательностей
В качестве родственной группы вирусов была выбрана группа, к которой принадлежит Karshi virus.
Геномы следующих вирусов лежат в этом fasta-файле:
Omsk hemorrhagic fever virus;
Phnom Penh bat virus strain 30834_A38;
Alkhurma virus;
Deer tick virus strain ctb30;
Negishi virus strain Negishi.
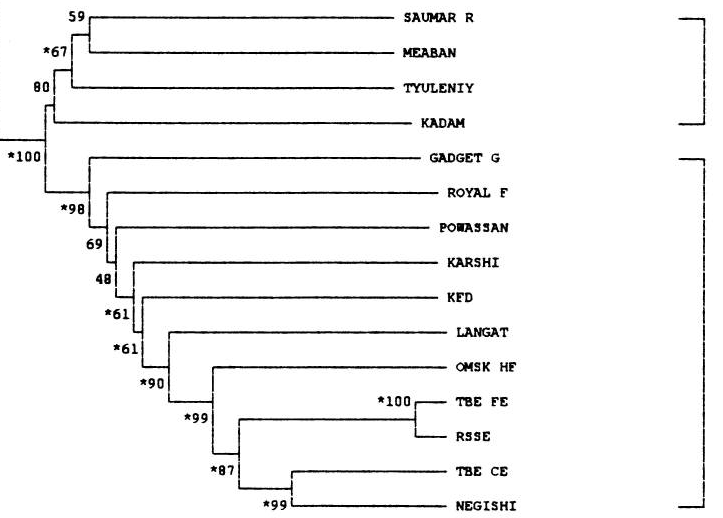
С помощью tblastx и скрипта на Python была составлена excel-таблица. В ней находятся 5 листов, отличающиеся по своему содержимому. Лист virus содержит исходную информацию, полученную при помощи выше описанных инструментов. Лист check содержит тот же список сравнений, что и virus, за исключением того, что он отсортирован по убыванию bit-score, а также включает проверку, заключающуюся в следующем: identity > 70%, bit-score > 700, длина выравнивания > 400 (А). После применения двух простых команд (написаны справа от таблицы) был создан еще один лист, results. После его анализа была составлена таблица наибольшего количества пересечений (таблица 1) и вынесена в отдельный лист, calc. Видно, что геномы вирусов с идентификаторами NC_005062.1 и KT224355.1, обладают наибольшим количеством пересечений, удовлетворяющих условию (А). Таким образом можно было бы предполагать, что эти вирусы наиболее близкородственны. Для того, чтобы это как-то подтвердить, был сделан еще один лист, pair, в котором представлены схемы нахождения среднего identity, а его результаты представлены в таблице 2 в листе calc. Видно, что наибольшее среднее identity как раз у ранее предсказанной пары. Кроме того, в этой статье строили деревья как раз для той группы вирусов, которую я взял в качестве исходной (Flavivirus). Вот части деревьев из той статьи:
 |
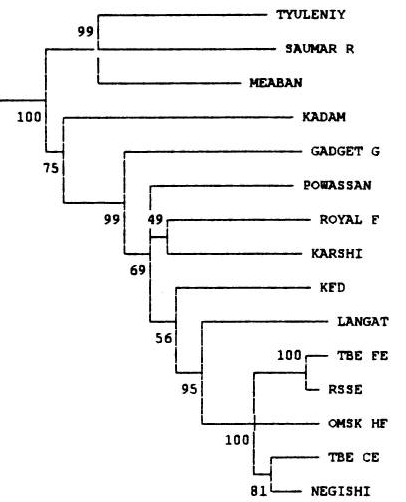 |
Видно, что вирусы Omsk hemorrhagic fever virus и Negishi virus strain Negishi достаточно близки, что является еще одним хорошим подтверждением правильности использования такого сравнения (identity > 70%, bit-score > 700, длина выравнивания > 400). Также, если проанализировать не настолько (11) хорошие пересечения, можно заметить, что организмы, чьи идентификаторы, отвечают заданному пересечению отстоят друг от друга на приличном расстоянии на деревьях выше.
⌘
© Emir Radkevich, 2016