EMBOSS
Упражнения по EMBOSS
Задача 1. Несколько файлов в формате fasta собрать в единый файл.
Исходные данные: первый файл, второй файл, третий файл с некоторыми последовательностями. Также все названия этих файлов были записаны в файл.
Команда в putty: {seqret -sequence @list.txt -outseq all.fasta}
Результат: ссылка на файл с собранными последовательностями.
Задача 2. Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы.
Исходные данные: исходный файл с тремя последовательностями.
Команда в putty: {seqretsplit -sequence all.fasta stdout}
Результат: первый файл, второй файл, третий файл.
Задача 3. Из файла с хромосомой в формате .gbk вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле.
Исходные данные: скачиваем три последовательности ниже (организм: Archangium gephyra).
Команда в putty: {echo -e
"genbank:CP011509[15541:15951]\ngenbank:CP011509[22828:23697:r]\ngenbank:CP011509[23801:24283]">cds.out}
{seqret -sequence @cds.out -outseq cds.fasta}
Результат: fasta файл с тремя последовательностями, кодирующими белки.
Задача 4. Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле.
Исходные данные: исходный файл с последовательностями, кодирующими белки.
Команда в putty: {transeq -table 0 -sequence cds.fasta -outseq allprot.fasta}
Результат: fasta файл с транслированными последовательностями.
Задача 5. Транслировать нуклеотидную последовательность в шести рамках.
Исходные данные: файл с тремя fasta-последовательностями.
Команда в putty: {transeq -frame 6 -sequence alltr.fasta -outseq alltr.pep}
Результат: на выходе шесть транслированных последовательностей для одной последовательности (ссылка на файл).
Задача 6. Перевести выравнивание из fasta формата в формат .msf.
Исходные данные: fasta файл с выравниванием последовательностей.
Команда в putty: {seqret -sequence align.fasta -outseq msf:align.msf}
Результат: msf файл с выровненными последовательностями.
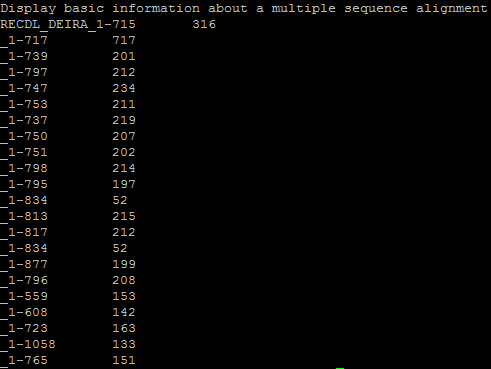
Задача 7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число).
Исходные данные: msf файл с выровненными последовательностями.
Команда в putty: {infoalign -refseq 2 -only -name -idcount -sequence align.msf stdout}
Результат: представлен на картинке снизу.
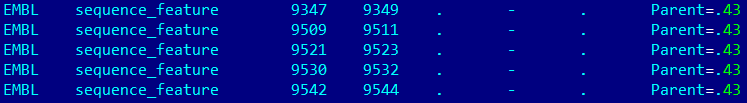
Задача 8. Перевести аннотации особенностей в записи формата .gbk в табличный формат .gff.
Исходные данные: gbk-файл с плазмидой.
Команда в putty: {featcopy -features NC_010466.gbk -outfeat NC_010466.gff}
Результат: часть таблицы можно увидеть на картинке снизу (ссылка на таблицу в формате .gff).
Задача 9. Из файла в формате .gbk получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product).
Исходные данные: gbk-файл с плазмидой.
Команда в putty: {extractfeat -type 'CDS' -describe 'product' -sequence NC_010466.gbk -outseq extr.fasta}
Результат: ссылка на fasta-последовательность.
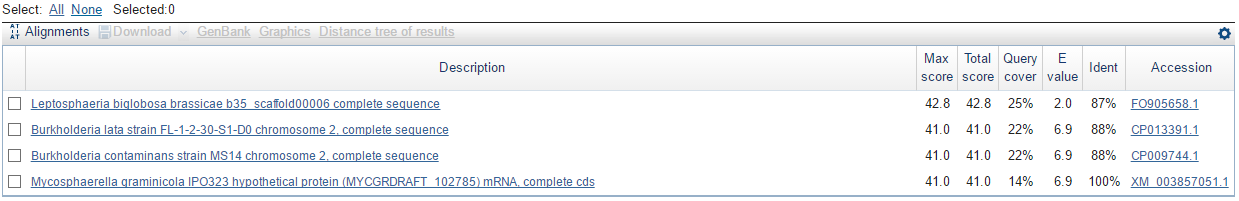
Задача 10. Перемешать буквы в нуклеотидной последовательности; (*) проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных (запустите с порогом E = 10 - по умолчанию).
Исходные данные: fasta-файл с некоторой последовательностью.
Команда в putty: {shuffle shuffle.fasta > sh.fasta}
Результат: ссылка на fasta-последовательность.
P.S.: алгоритм blastn выдал следующее: (видно, что 'достоверных' или 'около достоверных' находок нет)
Задача 11. Найти частоты кодонов в кодирующих последовательностях.
Исходные данные: fasta файл с последовательностями, кодирующими белки.
Команда в putty: {cusp -sequence cds.fasta -outfile cds.cusp}
Результат: cusp файл с посчитаными частотами кодонов.
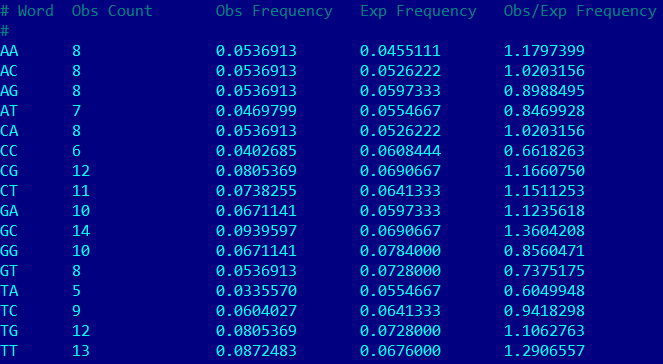
Задача 12. Найти частоты динуклеотидов в нуклеотидной последовательности и сравнить их с ожидаемыми.
Исходные данные: fasta-файл с некоторой последовательностью.
Команда в putty: {compseq -sequence sh.fasta -calcfreq -word 2 -outfile sh.composition}
Результат: ссылка на файл с посчитанными частотами. Также на картинке снизу часть выдачи программы (видно, что наблюдаемое не сильно отклоняется от ожидаемого).
Задача 13. Выровнять кодирующие последовательности соответственно выравниванию белков - их продуктов.
Исходные данные: fasta файл с последовательностями, кодирующие белки и fasta файл с выровненными белками.
Команда в putty: {tranalign -asequence cds.fasta -bsequence prali.fasta -outseq tranalign.fasta}
Результат: fasta файл с выровненными последовательностями.
Построение карты локального сходства
Были выбраны бактерии рода Mycobacterium: Mycobacterium intracellulare ATCC 13950, Mycobacterium africanum GM041182.
С помощью blast2seq была построена карта локального сходства (выбран word size 24, чтобы убрать шум):
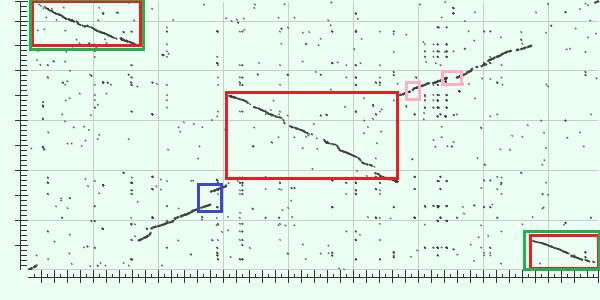
На OX отложена длина генома Mycobacterium africanum (4.389.314 bp), соответственно на OY - Mycobacterium intracellulare (5.402.402 bp). Сходство между гомологичными участками составляет 89%.
Красными прямоугольниками отмечены инверсии относительно Mycobacterium africanum (0.2Mb - 0.8Mb, 1.5Mb - 2.75Mb, 3.75Mb - 4.4Mb);
Зелеными прямоугольниками отмечены транслокации (0.2Mb - 0.8Mb и 3.75Mb - 4.4Mb);
Розовыми прямоугольниками отмечены делеции (2.95Mb, 3.25Mb);
Синим прямоугольником отмечена инсерция в геноме Mycobacterium intracellulare (1.4Mb).
⌘
© Emir Radkevich, 2016